U.S. FDA Accepts NDA and Grants Priority Review for Bayer’s Darolutamide in Non-Metastatic Castration-Resistant Prostate Cancer
Apr 30, 2019 09:41
CST Updated
09:41

Bayer
Pharmaceutical Product R&D Developer

FDA
U.S. Food and Drug Administration

April 30, 2019/BIOON/--German pharmaceutical giantBayer(Bayer) recently announced that the U.S. Food and Drug Administration (FDA) has accepted the New Drug Application (NDA) for darolutamide in the treatment of non-metastatic castration-resistant prostate cancer (nmCRPC) and granted it Priority Review. Previously,FDAFast-track designation was also granted for darolutamide in the treatment of nmCRPC. Bayer has recently submitted marketing authorization applications to the European Medicines Agency (EMA) and the Japanese Ministry of Health, Labour and Welfare (MHLW), and is also discussing submission-related matters with regulatory authorities in other countries.
This NDA submission andFDAPriority review was granted based on data from the pivotal Phase III ARAMIS clinical study. The results demonstrated that, in patients with non-metastatic castration-resistant prostate cancer (nmCRPC), darolutamide plus androgen deprivation therapy (ADT) significantly reduced the risk of metastasis or death by 59% compared with placebo plus ADT. Furthermore, the darolutamide plus ADT regimen exhibited a favorable safety profile relative to placebo plus ADT in this study.
Darolutamide is an oral non-steroidal androgen receptor (AR) antagonist with a unique chemical structure, exhibiting strong binding affinity to the receptor and potent antagonistic activity, thereby inhibiting receptor function and the growth of prostate cancer cells.
Darolutamide was co-developed by Bayer and the Finnish pharmaceutical company Orion. In addition to nmCRPC, the two parties are also advancing another Phase III clinical trial, ARASENS, to evaluate the efficacy and safety of darolutamide in the treatment of metastatic hormone-sensitive prostate cancer (mHSPC).
Senior Vice President of Bayer Pharmaceuticals Division andTumorScott Z. Fields, Head of Development, stated, “Bayer is committed to addressing the treatment gaps in the continuous care of patients with prostate cancer. With the acceptance of the New Drug Application (NDA) and priority review, we are one step closer to bringing darolutamide to patients as soon as possible.”
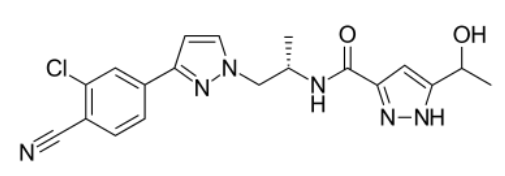
Darolutamide Molecular Structure (Image Source: Wikipedia)
ARAMIS was a randomized, multicenter, double-blind, placebo-controlled study that enrolled 1,509 patients who were receiving androgen deprivation therapy (ADT) as standard of care and were at high risk for metastatic disease. In the study, patients were randomized in a 2:1 ratio to receive either darolutamide (600 mg twice daily) or placebo, in addition to ADT. The primary endpoint was metastasis-free survival (MFS), defined as the time from randomization to evidence of metastasis or death. Secondary endpoints included overall survival (OS), time to pain progression, time to initiation of first cytotoxic chemotherapy, time to first symptomatic skeletal event (SSE), and the safety and tolerability profile of darolutamide.
The study results showed that, compared with the placebo + ADT group, the darolutamide + ADT group achieved a statistically significant improvement in MFS (HR = 0.41, 95% CI: 0.34–0.50, p < 0.001), indicating a 59% reduction in the risk of metastasis or death, thereby meeting the primary endpoint of the study. The median MFS was 18.4 months in the placebo + ADT group and 40.4 months in the darolutamide + ADT group, representing an overall prolongation of median MFS by 22 months.
In terms of overall survival (OS), the darolutamide + ADT regimen demonstrated a favorable trend compared with the placebo + ADT regimen, with a 29% reduction in the risk of death (HR=0.71, 95% CI: 0.50–0.99, p=0.045). Regarding progression-free survival (PFS), the darolutamide + ADT regimen significantly prolonged PFS compared with the placebo + ADT regimen (median PFS: 36.8 months vs. 14.8 months; HR=0.38, 95% CI: 0.32–0.45, p<0.001), reducing the risk of local progression, distant metastasis, or death by 62%.
Furthermore, all other secondary endpoints favored darolutamide, including time to pain progression (40.3 months vs. 25.4 months; HR=0.65, 95% CI: 0.53–0.79, p<0.001) and time to cytotoxic chemotherapy (not reached vs. 38.2 months; HR=0.43, 95% CI: 0.31–0.60, p<0.001). Another secondary endpoint, time to first symptomatic skeletal event (SSE), also favored darolutamide.
Importantly, the incidence of treatment-emergent adverse events (AEs) occurring in ≥5% of patients or classified as Grade 3–5 was comparable between the two treatment groups; only fatigue occurred in more than 10% of patients (12.1% in the darolutamide + ADT group vs. 8.7% in the placebo + ADT group), and quality-of-life outcomes were similar between the two treatment groups.
These data were presented in mid-February this year at the American Society of Clinical Oncology Genitourinary Cancers Symposium held in San Francisco, United States.Tumorpresented at the ASCO GU Symposium and simultaneously published in The New England Journal of Medicine (NEJM). (Bioon.com)