

The remarkable success of immune checkpoint inhibitors (ICBs), such as PD-1/PD-L1 antibodies, has refocused the attention of the scientific and industrial communities on harnessing the immune system to eliminate cancer cells.
Immune checkpoint blockade (ICB) restores anti-tumor immune responses by blocking immunosuppressive tumor signals through targeting receptor or ligand checkpoint proteins, such as PD-1/PD-L1 and CTLA-4. Numerous trials have demonstrated that ICB antibodies, whether used as monotherapy or in combination regimens, significantly improve objective response rates and survival outcomes compared to standard therapies for various solid tumors. However, it is important to note that the majority of patients do not respond to ICB. Furthermore, the factors governing successful immune-mediated tumor cell elimination during ICB therapy are not yet fully understood, and scientists continue to investigate definitive biomarkers capable of predicting tumor response to ICB.
However, evidence indicates that immune checkpoint blockade (ICB) confers the greatest benefit, i.e., the strongest anticancer effect, in tumors with high levels of tumor-infiltrating lymphocytes (TILs), high mutational burden, and increased PD-L1 expression. These responsive tumors are referred to as immunologically “hot tumors,” in contrast to non-responsive “cold tumors.” Studies have shown that the lack of response to ICB therapy in these “cold tumors” is primarily attributed to the lack of expression and/or presentation of tumor-associated antigens (TAAs), low density of TILs, infiltration by inhibitory immune cell subsets (such as neutrophils, macrophages, regulatory T cells, myeloid-derived suppressor cells, and natural killer cells), and the expression of immunosuppressive substances (such as IL-10, indoleamine-2,3-dioxygenase, CD73, PD-L1, and prostaglandin E2).
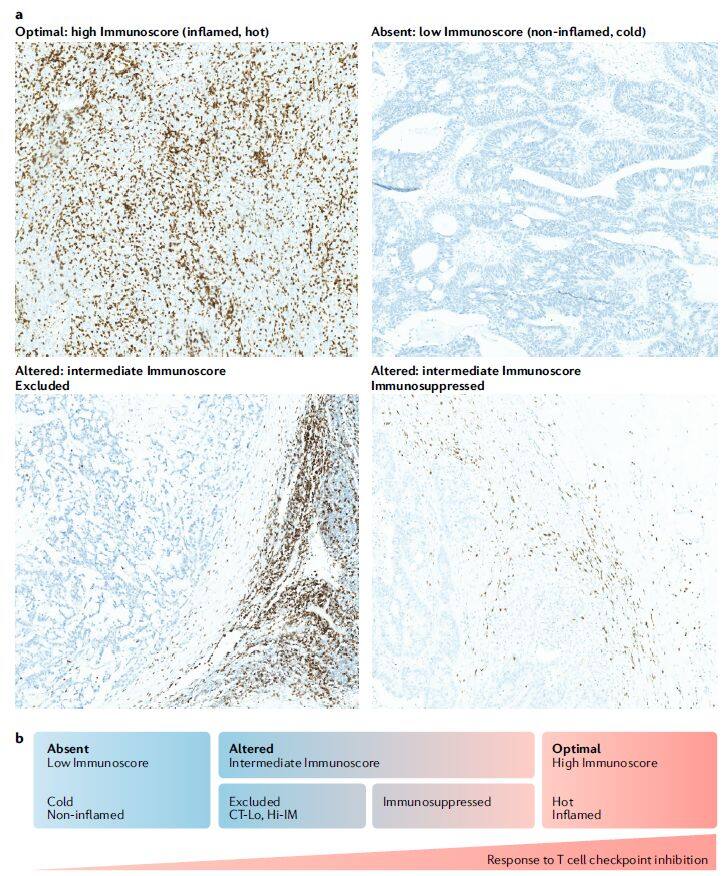
Definition of Cold and Hot Tumors (Image source: Nature Reviews Drug Discovery[1])
Although emerging data have broadened our understanding of tumor biology and ICB-induced anti-tumor immune responses, second-generation immune-modifying agents aimed at restoring T cell anti-cancer functions have not provided the reproducible and durable clinical benefits expected based on preclinical studies. These second-generation immune-modifying agents include agonist antibodies targeting members of the tumor necrosis factor receptor family (such as OX40, 4-1BB, GITR), as well as compounds targeting immunosuppressive pathways in the tumor microenvironment (TME) (such as indoleamine-2,3-dioxygenase, arginase 1).
In light of these current realities, the industry has begun seeking next-generation therapeutic strategies to address resistance to immune checkpoint blockade (ICB), with the aim of creating immunologically “hot tumors.” Among these, oncolytic viruses (a class of viruses that preferentially infect and kill cancer cells) represent an ideal therapeutic approach for enhancing patient response to ICB in certain tumor types, owing to their ability to self-replicate and mediate antitumor activity through multiple mechanisms. Although much remains to be understood about how oncolytic viruses can be most effectively combined with ICB, they can be employed to promote lymphocyte recruitment, induce PD-1/PD-L1 expression to increase tumor responsiveness to ICB and reverse resistance, and additionally modulate other components of the antitumor immune response.
Accumulating research indicates that the mechanisms of action of oncolytic viruses include direct tumor lysis, modulation of the tumor microenvironment, recruitment of tumor-infiltrating lymphocytes (TILs), initiation of immune responses mediated by CD8+ T cells and innate immune cells, and regulation of vasculature (e.g., inhibition of tumor angiogenesis). Oncolytic viruses can also promote immunogenic cell death, which further leads to the release of danger-associated molecular patterns (DAMPs), thereby attracting innate immune cells (particularly dendritic cells) into the tumor and resulting in the recruitment and maturation of tumor-specific T cells within the tumor microenvironment.
Oncolytic viruses can selectively infect and kill tumor cells through natural mechanisms, such as by enhancing the sensitivity of tumor cells to antiviral/antimicrobial signaling pathways (e.g., those mediated by cytosolic nucleic acid sensors including cGAS-STING, RIG-I, MDA5, and Toll-like receptors). Furthermore, their host range can be restricted to selectively target tumors by directly genetically engineering oncolytic viruses to delete genes required for replication in normal cells but dispensable for replication in tumor cells.
However, although the tumor-selective targeting property of a series of armed and non-armed oncolytic viruses has been validated, their efficacy as monotherapy remains limited in both preclinical and clinical settings.
To date, only one oncolytic virus has been approved for marketing in both the United States and Europe: T-VEC, developed by Amgen for the treatment of advanced melanoma. T-VEC is an oncolytic virus based on herpes simplex virus type 1 (HSV-1), engineered to preferentially target tumor cells. This specificity is achieved by deleting the RL1 gene, which encodes ICP34.5; the absence of ICP34.5 prevents viral replication in normal cells. Additionally, the developers have enhanced the virus to express human granulocyte-macrophage colony-stimulating factor (GM-CSF) to stimulate or augment anti-tumor immune responses.
The approval of T-VEC, which intersects virology, cancer biology, and immunology, has significantly propelled the development of this class of oncolytic viruses. Extensive research has demonstrated that arming these viruses with transgenes capable of enhancing their immune-mediated activity or inducing novel immunomodulatory patterns within the tumor microenvironment holds promise for substantially improving tumor responses to immune checkpoint blockade (ICB), particularly in immunologically “cold” tumors. The limited toxicity of oncolytic viruses and their ability to modulate the tumor microenvironment provide compelling opportunities for combination with other anticancer therapies featuring complementary mechanisms of action.

Image source: Nature Reviews Drug Discovery
On July 10, in a review article published in Nature Reviews Drug Discovery [2], five scientists from the Institute of Cancer Research and AstraZeneca’s Oncology R&D division discussed the development of oncolytic virus therapies, aiming to optimize their ability to create “hot tumors” and to guide future combination treatments.
I. Virus Selection
Given that most oncolytic viruses lack durable responses as monotherapies, current industry research efforts are focused on enhancing the antitumor efficacy of oncolytic viruses. Selecting an oncolytic virus and its combination partner requires consideration of multiple factors (Figure 1).
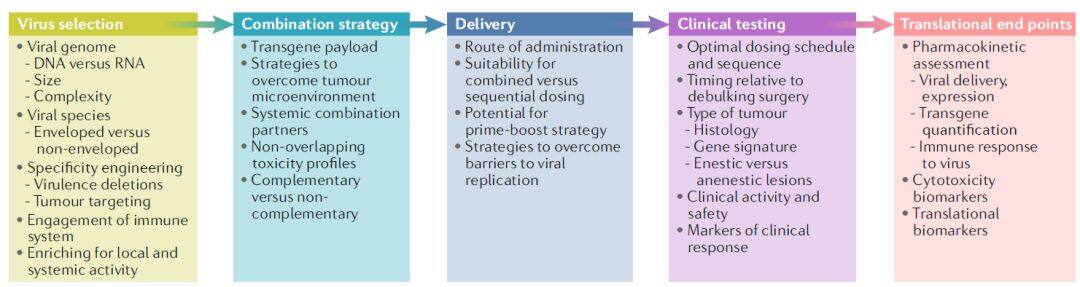
Figure 1 | Oncolytic viruses as part of the tumor immunotherapy toolbox (Image source: Nature Reviews Drug Discovery)
The genomes of oncolytic viruses can consist of single-stranded or double-stranded RNA or DNA, exhibiting diverse structures, genomic organizations, expression strategies, and replication mechanisms (Table 1). Furthermore, viral families may differ in their lytic capacity, ability to induce innate and adaptive immune responses, and capability to package transgenes. Although many oncolytic viruses have been genetically engineered to achieve tumor specificity, some unmodified oncolytic viruses have also entered clinical trials, such as reovirus, coxsackievirus A21, and HF10 (Table 2).
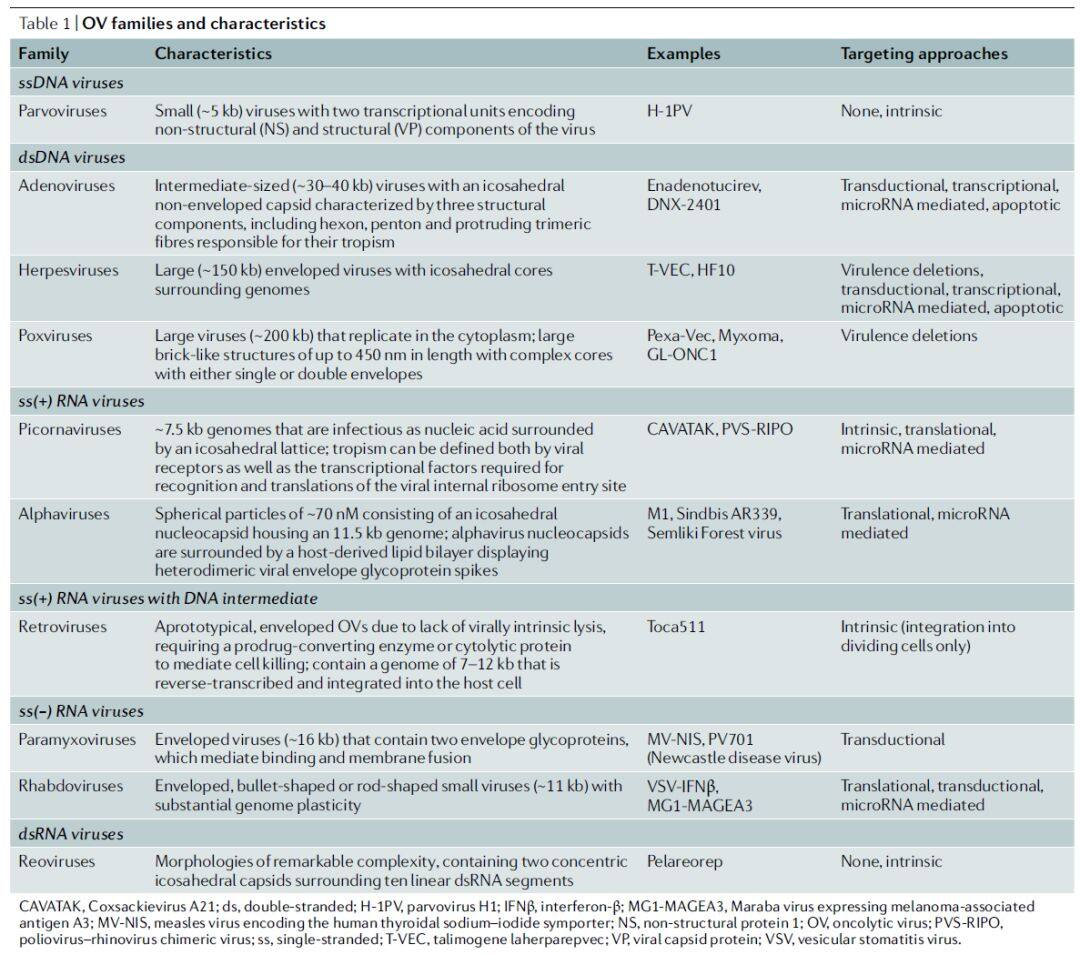
Table 1 | Oncolytic virus families and their characteristics (Data source: Nature Reviews Drug Discovery)
On the other hand, although the gene and protein expression profiles of tumor cells may provide a certain degree of inherent oncolytic selectivity, many approaches have been employed to further enhance the tumor specificity of oncolytic viruses.
Most viruses can be engineered to encode exogenous genes. However, RNA viruses typically have smaller genomes and more limited packaging capacity, whereas DNA viruses (such as HSV and vaccinia virus) generally possess larger genomes, are easier to manipulate without compromising viral replication, and can accommodate larger transgenes or multiple genes. Furthermore, owing to high-fidelity DNA polymerases, DNA viruses may exhibit better genomic integrity and potentially represent more stable therapeutic products.
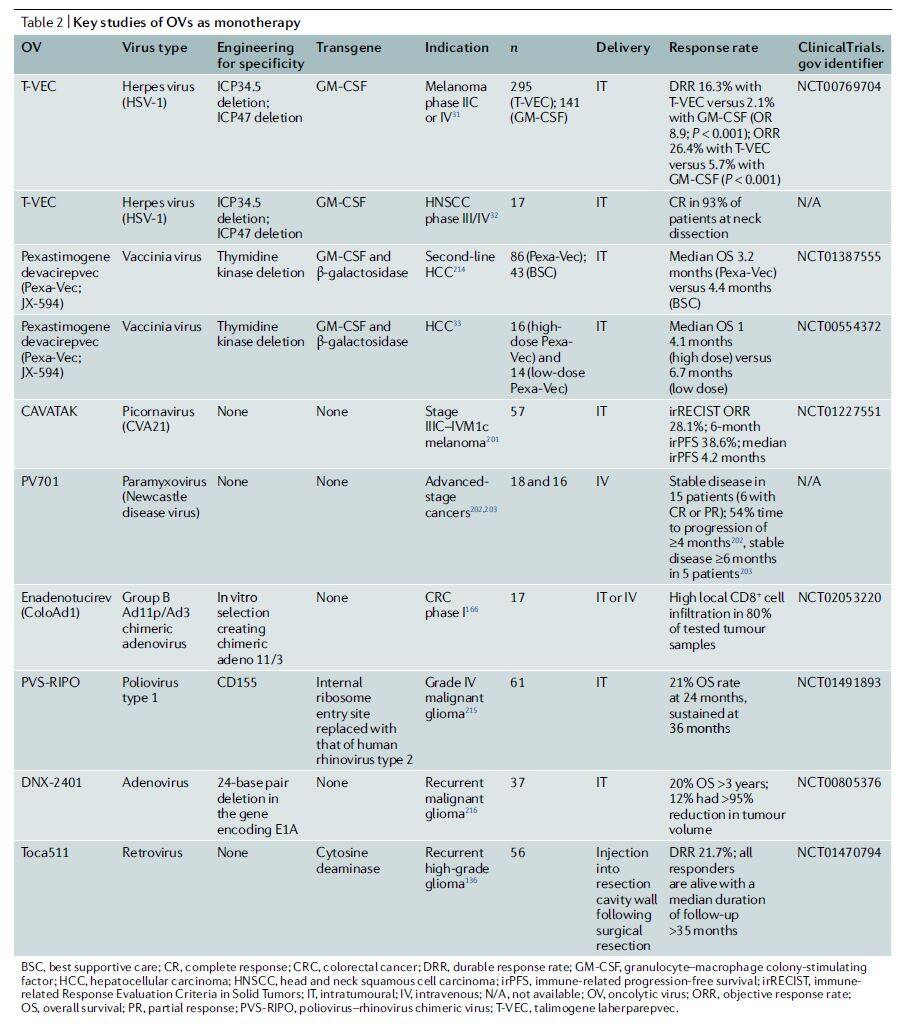
Table 2 | Key Studies of Oncolytic Viruses as Monotherapy (Data Source: Nature Reviews Drug Discovery)
II. Mechanism of Action
Although some studies have confirmed that oncolytic viruses exert their antitumor effects through direct tumor lysis and indirect induction of systemic antitumor immunity, the relative importance of each mechanism to overall therapeutic efficacy remains unclear. Through extensive research on numerous wild-type and genetically engineered viruses, an increasing number of safe and effective oncolytic viruses have been developed; these viruses exhibit varying degrees of tumor tropism, lytic capacity, ability to induce innate and adaptive immune responses, and packaging capacity.
In addition to tumor selectivity, another important consideration in the genetic engineering of oncolytic viruses is to find an optimal balance between lytic potential and the ability to stimulate anti-tumor adaptive immune responses, ensuring that the tumor is not lysed before producing immunomodulatory molecules that create a more favorable tumor microenvironment.
Before deciding to arm oncolytic viruses with transgenes or combine them with other therapies, numerous factors beyond the virus’s inherent oncolytic and immunomodulatory capacities must be considered, including the site of action, treatment duration, and cost. Further analysis of primary tumor samples will provide deeper insights into baseline immune infiltration within the tumor microenvironment and may guide armed oncolytic virus strategies or rational combination therapies in the future (Figure 2).
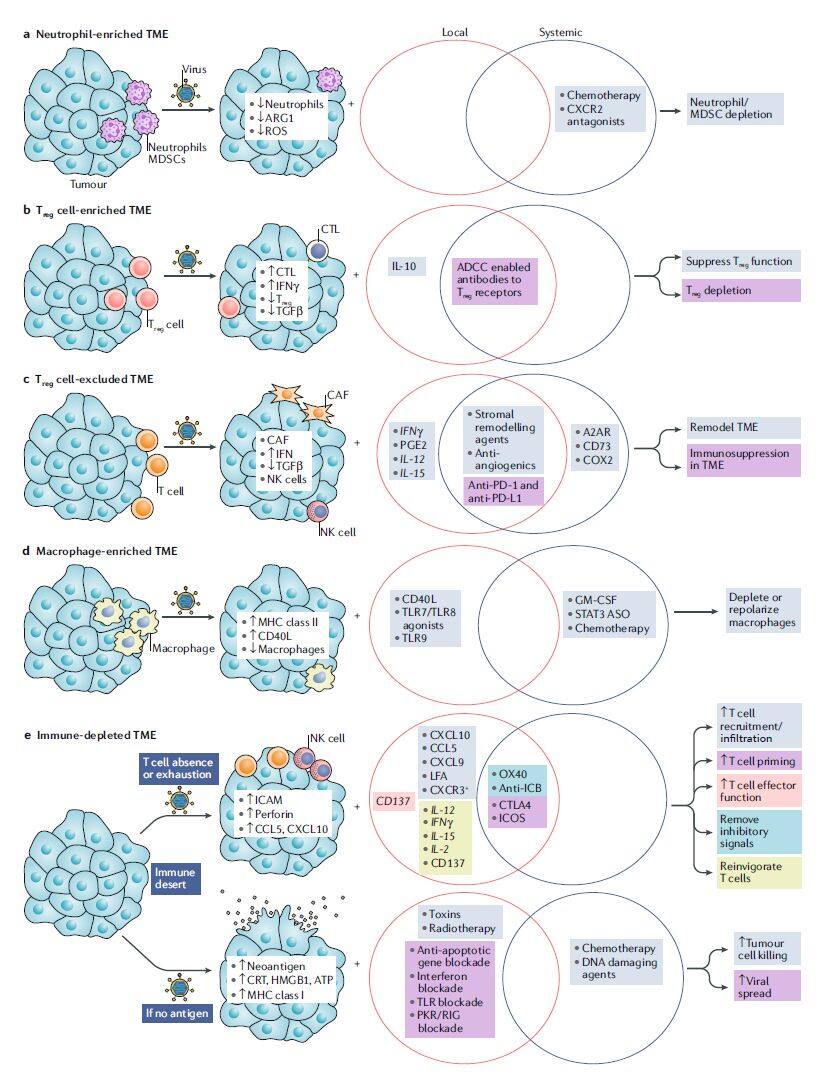
Figure 2 | Designing local and systemic combination strategies to enhance the activity of oncolytic viruses (Image source: Nature Reviews Drug Discovery)
III. Combination Strategy
The most rapidly advancing oncolytic virus combination regimen in clinical practice is the combination with PD-1/PD-L1 antibodies (Table 3), with preliminary data showing promising prospects [3]. Combination regimens of oncolytic viruses with CTLA-4, TIM3, or LAG3 antibodies are also under evaluation. Furthermore, oncolytic viruses armed with multiple cytokines have been used in combination with CAR-T cell therapy; in xenograft tumor models, this combination strategy enhanced antitumor activity [4].
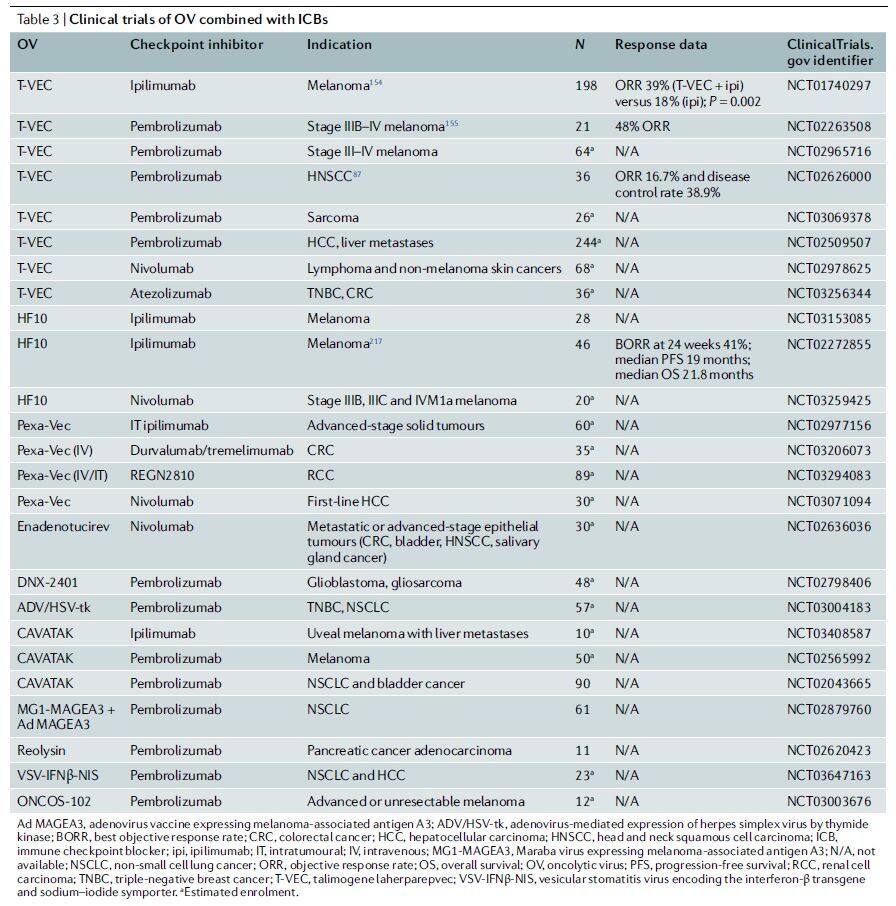
Table 3 | Clinical Trials of Oncolytic Viruses Combined with ICB (Data Source: Nature Reviews Drug Discovery)
IV. Armament Strategy
To drive a robust adaptive immune response, engineering oncolytic viruses to express bispecific T-cell engagers (targeting CD3 and tumor-associated antigens [TAAs]) represents a viable arming strategy. Studies have confirmed [5-6] that such oncolytic viruses demonstrate oncolytic activity in primary ex vivo patient samples and in vivo xenograft models, as well as immune-mediated antitumor effects resulting from the recruitment and activation of cytotoxic T cells.
V. Overcoming In Vivo Response Barriers
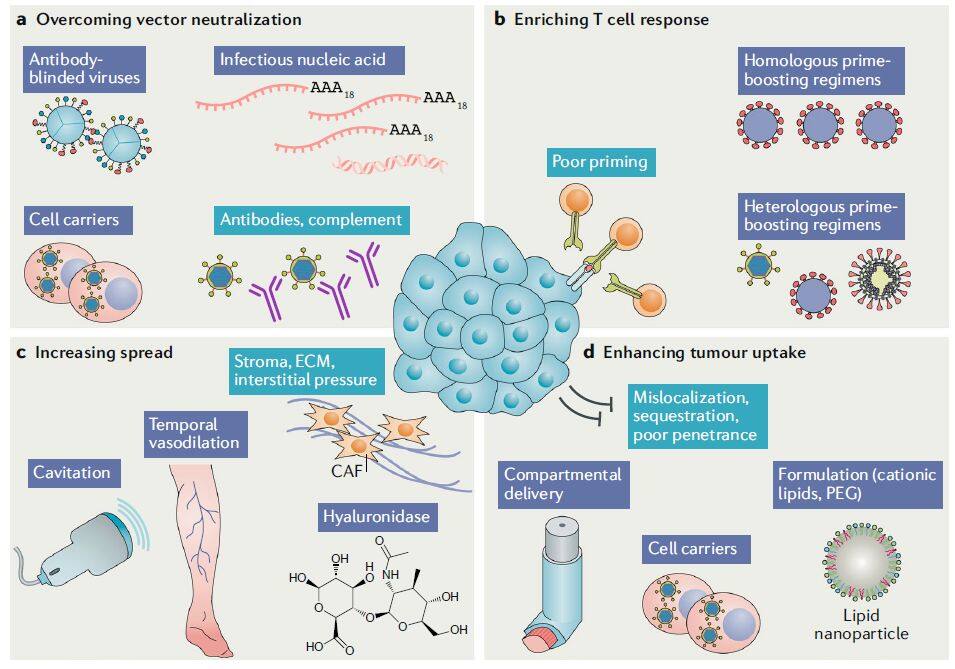
Figure 3 | Overcoming in vivo response barriers (Image source: Nature Reviews Drug Discovery)
In addition to arming and combination strategies designed to enhance targeting and efficacy, other approaches can also be employed to overcome in vivo barriers to oncolytic virus delivery (Figure 3). The presence of antivector antibodies hinders the systemic administration of many oncolytic viruses. To address this challenge, some researchers have proposed a strategy of using cell carriers to allow oncolytic viruses to “hitchhike”; these cells exhibit tropism for tumor tissues, thereby shielding the viruses. For instance, clinical studies have demonstrated that peripheral blood mononuclear cells can serve as carriers for oncolytic viruses.
VI. Delivery Route
The efficacy of oncolytic viruses depends on infecting target tumor cells with a sufficient quantity of the virus, leading to cell lysis and the spread of the oncolytic virus to adjacent tumor cells. The dose-response relationship of oncolytic viruses is difficult to predict, partly due to their self-replicating capability. Although most studies to date have employed local administration (intratumoral, intraperitoneal, or intracranial) (Table 4), various delivery methods for oncolytic viruses are currently under investigation. Ultimately, effective treatment of advanced metastatic disease may require systemic administration.
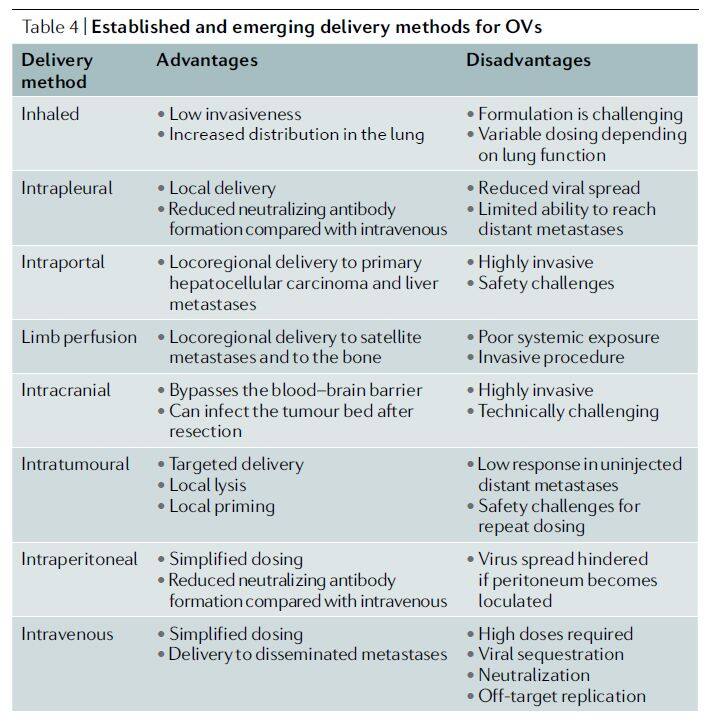
Table 4 | Existing and emerging delivery methods for oncolytic viruses (Source: Nature Reviews Drug Discovery)
1. Intratumoral Delivery
Intratumoral delivery is currently the most common route of administration for oncolytic viruses. Its most significant advantage lies in facilitating direct viral entry into the tumor, thereby avoiding systemic dilution, attack by anti-vector immunity, and sequestration at off-target sites, which reduces the risk of replication in non-target cells. Notably, studies have confirmed [7] that although oncolytic effects based on this delivery strategy may be confined to the tumor sites injected with the oncolytic virus, antitumor immune responses can also occur in uninjected lesion areas when a sufficient systemic immune response is induced.
2. Intravenous Injection
Intravenous delivery is a simpler and more favored route of administration among oncologists, as it allows drugs to reach multiple metastatic sites and aligns with the standard practices for administering many cancer therapies. Furthermore, intravenous infusion of oncolytic viruses offers significant convenience and safety benefits. However, some studies indicate that intravenous injection of oncolytic viruses has not yet been successful, as factors such as dilution, rapid clearance by neutralizing antibodies, accumulation in non-target organs, and/or the inability of the virus to extravasate through tumor vasculature necessitate the use of higher viral particle doses.
3. Intraperitoneal Delivery
Intraperitoneal delivery enables viruses to localize within larger body cavities. Although, similar to intravenous administration, the intraperitoneal route may also lead to poor distribution of oncolytic viruses and rapid viral clearance, a previous study [8] has shown that intraperitoneal delivery restricts the systemic biodistribution of viruses more effectively than intravenous therapy, resulting in significantly longer overall survival.
4. Other Delivery Routes
Other routes for delivering oncolytic viruses also include isolated limb perfusion, intracranial delivery, aerosol delivery, intraosseous delivery, intra-arterial infusion, portal vein delivery, and ultrasound cavitation (Figure 3).
VII. Efficacy Assessment
Biomarkers (such as viral DNA or proteins) or imaging techniques are expected to be used to evaluate the efficacy of oncolytic viruses, providing information on whether the oncolytic virus has reached the target tumor site, the extent of tumor cell killing, and whether the oncolytic virus has modulated the tumor microenvironment (e.g., converting “cold” tumors into “hot” tumors).
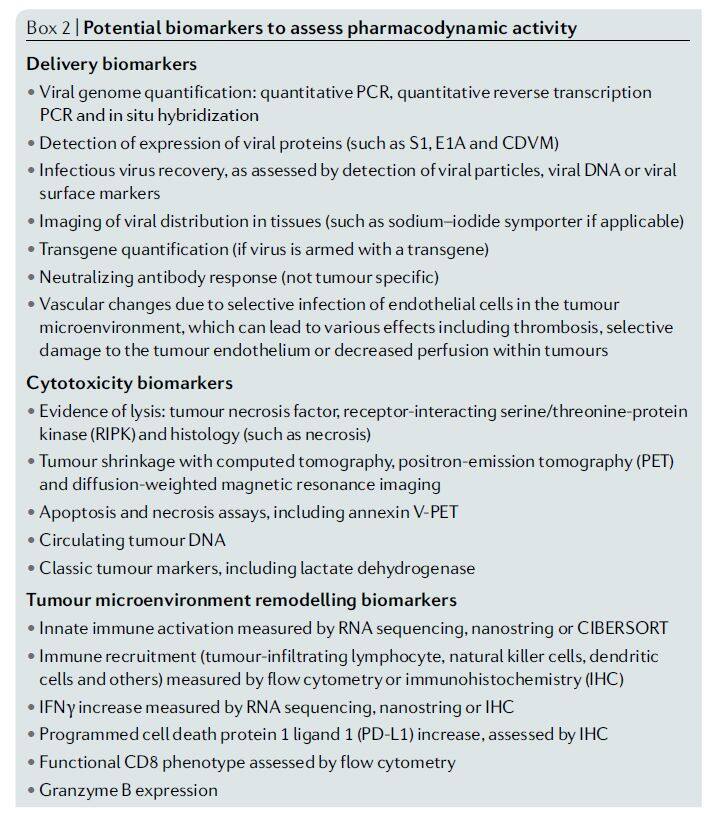
Figure 4 | Potential biomarkers for assessing pharmacodynamic activity (Image source: Nature Reviews Drug Discovery)
VIII. Summary
To date, the development of oncolytic viruses has spanned more than a century. This field has progressed from initial research involving the direct use of wild-type viruses, to the selective engineering of viral genomes to enable tumor-specific replication, and further to the incorporation of various exogenous genes into oncolytic viruses to enhance their therapeutic efficacy, achieving substantial advancements in this therapeutic approach.
In recent years, the successful launch of T-VEC and the prevalence of resistance to immune checkpoint blockade (ICB) therapy in most patients have propelled the development of oncolytic viruses to a new "peak." Scientists increasingly recognize that oncolytic viruses possess unique immunomodulatory effects, holding promise for enhancing the therapeutic potential of ICB. Currently, multiple trials are evaluating combination strategies involving oncolytic viruses plus ICB. Whether oncolytic viruses can truly "add fuel to the fire" of cancer immunotherapy awaits further validation through additional data.
Related Papers:
[1] Jérôme Galon et al. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nature Reviews Drug Discovery(2019).Jérôme Galon
[2] Kevin Harrington et al. Optimizing oncolytic virotherapy in cancer treatment. Nature Reviews Drug Discovery(2019).
[3] Christopher J. LaRocca et al.Oncolytic viruses and checkpoint inhibitors: combination therapy in clinical trials. Clinical and Translational Medicine(2018).
[4] Kiyonori Tanoue et al. Armed Oncolytic Adenovirus–Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Research(2017).
[5] Carlos Alberto Fajardo et al. Oncolytic Adenoviral Delivery of an EGFR-Targeting T-cell Engager Improves Antitumor Efficacy. Cancer Research(2017).
[6] Joshua D Freedman et al. Oncolytic adenovirus expressing bispecific antibody targets T‐cell cytotoxicity in cancer biopsies. EMBO Molecular Medicine(2017).
[7] Robert H. I. Andtbacka etal. Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Annals of Surgical Oncology(2016).
[8] Y Kulu et al. Comparison of intravenousversus intraperitoneal administration of oncolytic herpes simplex virus 1 for peritoneal carcinomatosis in mice. Cancer Gene Therapy(2009).