Roche's Risdiplam, the First Oral Therapy for Spinal Muscular Atrophy, Demonstrates Success in Pivotal FIREFISH Trial for Type 1 SMA
Jan 24, 2020 11:19
CST Updated
11:19

Roche
Oncology Drug Research, Development, and Manufacturing

January 24, 2020 /BioValleyBIOON/ -- Swiss pharmaceutical giant Roche recently announced positive results from Part 2 of the pivotal FIREFISH study (NCT02913482), which evaluated risdiplam for the treatment of infants with Type 1 spinal muscular atrophy (SMA).Risdiplam is a splicing modifier of the survival motor neuron 2 (SMN2) gene, developed for the treatment of all types (Type 1, Type 2, and Type 3) of spinal muscular atrophy (SMA). Currently, the drug is under priority review by the U.S. Food and Drug Administration (FDA), with a decision expected in May this year. If approved,Risdiplam will become the first oral medication for the treatment of all three types of SMA.
As part of its collaboration with the SMA Foundation and PTC Therapeutics, Roche led the clinical development program for risdiplam. If approved, Roche will be responsible for the commercialization of risdiplam in the United States.
FIREFISH is an open-label, multicenter Phase II/III study evaluating the safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and efficacy of risdiplam in infants aged 1 to 7 months with Type 1 spinal muscular atrophy (SMA). The study comprises two parts: an exploratory dose-finding part (Part 1, n=21) and a confirmatory part (Part 2, n=41). In Part 2, patients will receive oral risdiplam at the dose selected in Part 1 for 24 months. The primary endpoint is the proportion of infants able to sit without support for at least 5 seconds after 12 months of treatment, as assessed by the Motor Scale of the Bayley Scales of Infant and Toddler Development, Third Edition (BSID-III).
The results showed that the study met its primary endpoint: infants with type 1 SMA treated with risdiplam demonstrated statistically and clinically significant improvements in motor milestones. In this study, the safety profile of risdiplam was consistent with its known safety profile, and no new safety signals were identified.
Data from this study will be presented at the upcoming medicalMeetingpublished above. To date, more than 400 patients have received risdiplam treatment across all studies, and no treatment-related safety findings leading to discontinuation have occurred in any study.
Levi Garraway, MD, Chief Medical Officer and Global Head of Product Development at Roche, stated: “This large-scale global trial confirms the efficacy of risdiplam in advanced and refractory populations, including many infants whose disease had already progressed significantly before treatment initiation. We are highly encouraged by these results and look forward to sharing them with regulatory authorities. We also thank the entire SMA community for their continued collaboration.”
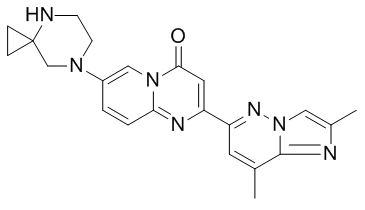
Chemical Structure of Risdiplam (Image source: medchemexpress.cn)
Risdiplam is an oral liquid and a splicing modifier of the survival motor neuron 2 (SMN2) gene, designed to continuously increase and maintain SMN protein levels in the central nervous system and peripheral tissues. Growing clinical evidence indicates that SMA is a multisystem disease, and the loss of SMA protein may affect many tissues and cells beyond the central nervous system. After oral administration, risdiplam exhibits systemic distribution, sustainably increasing SMN protein levels in both the central nervous system and peripheral tissues, and has been shown to improve motor function in patients with type 1, type 2, and type 3 SMA. Risdiplam is poised to become the first oral medication for the treatment of all three types of SMA.
In November 2019, the U.S. FDA accepted the New Drug Application (NDA) for risdiplam in the treatment of spinal muscular atrophy (SMA) and granted it Priority Review, with a Prescription Drug User Fee Act (PDUFA) target action date of May 24, 2020. Previously,FDARisdiplam has been granted orphan drug designation and fast track designation. Risdiplam is an oral liquid formulation; if approved, it will become the first medication available for home administration to patients with spinal muscular atrophy (SMA).
The risdiplam NDA includes 12-month data from Part 1 (dose-finding) of the pivotal FIREFISH and SUNFISH studies, as well as data from Part 2 (confirmatory) of the SUNFISH study.
SUNFISH is a two-part, double-blind, placebo-controlled pivotalClinical Trial, conducted in children and young adults (aged 2–25 years) with type 2 or type 3 spinal muscular atrophy (SMA). Part 1 determined the dose for the confirmatory Part 2, with exploratory endpoints assessing efficacy. Part 2 was a large placebo-controlled trial evaluating risdiplam in patients with type 2 or type 3 SMA. Data from Part 2, released in November 2019, showed that the study met its primary endpoint of change from baseline in the Movement Function Measure-32 (MFM-32) scale score: after one year of treatment, patients receiving risdiplam demonstrated significant improvements in motor function compared with the placebo group.
In addition to the studies included in the NDA, risdiplam is being investigated in a broad SMAClinical TrialResearch conducted within the project enrolled patients ranging from neonates to adults up to 60 years of age, including those who had previously received SMA therapy. Currently, Roche is conducting four global, multicenter clinical trials (SUNFISH [NCT02908685], FIREFISH [NCT02913482], JEWELFISH [NCT03032172], and RAINBOWFISH [NCT03779334]) to evaluate the efficacy and safety of risdiplam in treating all types (Type 1, Type 2, and Type 3) of SMA, as well as presymptomatic SMA in neonates.
Spinal Muscular Atrophy (SMA) is a motor neuron disease that causes muscle weakness and atrophy. It is an autosomal recessive genetic disorder caused by gene defects, affecting muscles throughout the patient's body. Patients primarily present with generalized muscle wasting and weakness, progressively losing various motor functions, including breathing and swallowing. SMA is the leading cause of death in infants under two years of age.GeneticsSpinal Muscular Atrophy (SMA) is a relatively common "rare disease," with an incidence rate of 1 in 6,000 to 1 in 10,000 among newborns. According to relevant reports, there are currently approximately 30,000 to 50,000 SMA patients in China.
In December 2016, Spinraza (nusinersen), a drug developed by Biogen and its partner Ionis, was approved, becoming the first therapy worldwide for spinal muscular atrophy (SMA). This medication is an antisense oligonucleotide (ASO) administered via intrathecal injection, delivering the drug directly into the cerebrospinal fluid (CSF) surrounding the spinal cord. It modifies the splicing of SMN2 pre-messenger RNA (pre-mRNA), thereby increasing the production of full-length functional SMN protein. In patients with SMA, insufficient levels of SMN protein lead to the degeneration of motor neuron function in the spinal cord. Clinical studies have demonstrated that treatment with Spinraza significantly improves motor function in patients with SMA.
In May 2019, fromNovartisThe gene therapy Zolgensma (onasemnogene abeparvovec) was approved, becoming the world’s first gene therapy for the treatment of spinal muscular atrophy (SMA). Administered as a single, one-time intravenous infusion, the drug enables sustained expression of the SMN protein to halt disease progression, addresses the root cause of SMA, and holds promise for long-term improvement in patients’ quality of life.
In the Chinese market, Spinraza was approved in late February 2019 for the treatment of patients with 5q spinal muscular atrophy (5q-SMA). This approval made Spinraza the first drug for treating SMA in China. 5q-SMA is the most common type of SMA, accounting for approximately 95% of all SMA cases. This form of SMA is caused by mutations in the SMN1 (survival motor neuron 1) gene on chromosome 5, hence the name 5q-SMA. (Bioon.com)