Pfizer’s Vyndaqel (tafamidis) Approved by European Commission as First Treatment for Transthyretin Amyloid Cardiomyopathy (ATTR-CM)
Feb 19, 2020 13:10
CST Updated
13:10

Pfizer
Pharmaceutical R&D Developer

European Commission
The European Commission, abbreviated as the EU Commission, is a supranational body under the European Union. Within the EU political system, the European Commission primarily undertakes executive tasks, thus being roughly equivalent to the government in a national system. However, the European Commission has other functions as well. In particular, except for the few circumstances specified in the treaties, the European Commission is the only institution with legislative power in the EU legislative process.

February 19, 2020 / Bioon -- Pfizer recently announced that the European Commission (EC) has approved Vyndaqel (tafamidis), a once-daily 61 mg oral capsule for the treatment of adult patients with wild-type or hereditary transthyretin-mediated amyloid cardiomyopathy (ATTR-CM). Prior to this approval, treatment options for patients with ATTR-CM were limited to symptom management and, in rare cases, heart (or combined heart and liver) transplantation. ATTR-CM is a rare, life-threatening disease characterized by the accumulation of misfolded proteins known as amyloids in the heart, defined by restrictive cardiomyopathy and progressive heart failure. On average, patients survive only 2 to 3.5 years after diagnosis.
In 2011, the European Union approved a different formulation of Vyndaqel (tafamidis meglumine) 20 mg capsules for the treatment of transthyretin amyloidosis in adult patients with stage 1 symptomatic polyneuropathy (ATTR-PN) to delay impairment of peripheral neurological function. For ATTR-CM, the tafamidis 61 mg capsule is equivalent to an 80 mg dose of tafamidis meglumine (four 20 mg capsules). This capsule was developed to enhance patient convenience by allowing for once-daily dosing. Although both products contain the same active ingredient, tafamidis, they are not interchangeable due to differences in recommended dosing.
It is worth mentioning that,Vyndaqel is the first and only drug approved in the EU for the treatment of ATTR-CM、The only drug proven to reduce mortality and cardiovascular-related hospitalization rates in patients with wild-type or hereditary ATTR-CM. In the European Union, Vyndaqel is alsoThe first drug capable of simultaneously treating ATTR-CM and Stage 1 symptomatic transthyretin amyloidosis with polyneuropathy (ATTR-PN)。
In the United States, Vyndaqel (tafamidis meglumine) and Vyndamax (tafamidis) were approved in early May 2019FDAApproved as the first and only drug for the treatment of ATTR-CM. The two drugs are two oral formulations of tafamidis, a first-in-class transthyretin stabilizer.
Paul Levesque, Global President of Pfizer Rare Disease, stated: “Previously, there were no medicines approved in the EU for the treatment of patients with ATTR-CM. Today’s approval represents a significant milestone for these patients, reflecting our unwavering commitment to delivering breakthrough medicines to those living with rare diseases. Furthermore, with today’s milestone, Vyndaqel is now the first medicine approved in the EU with two formulations for the treatment of transthyretin amyloidosis manifestations: one for cardiomyopathy and one for Stage 1 polyneuropathy.”
Thibaud Damy, former chair of the Heart Failure and Cardiomyopathy Working Group of the French Society of Cardiology and coordinator of the French Referral Center for Cardiac Amyloidosis, stated: “Prior to today, there was an urgent need for new treatment options within the European transthyretin amyloidosis community to improve outcomes in patients with cardiomyopathy. Vyndaqel represents a significant advance for patients, as it can significantly reduce wild-type orHeredityall-cause mortality and frequency of cardiovascular-related hospitalizations in patients with ATTR-CM.”
The European Commission approved Vyndaqel based on data from the pivotal Phase III ATTR-ACT clinical study, which is the first and only successfully completed global, double-blind, randomized, placebo-controlled trial investigating a pharmacological therapy for ATTR-CM.Clinical Trials. This study enrolled a total of 441 patients, including those with variant or hereditary ATTR-CM and those with wild-type ATTR-CM (referring to those who are notGenetics, but rather may occur with advancing age).
The primary analysis results showed that during the 30-month treatment period: (1) Compared with placebo, Vyndaqel (tafamidis meglumine) significantly reduced all-cause mortality and cardiovascular-related hospitalization rates in patients with wild-type and hereditary ATTR-CM (p=0.0006). (2) Compared with the placebo group, Vyndaqel reduced all-cause mortality by 30% (p=0.026) and all-cause hospitalization rate by 32% (p<0.0001). (3) Compared with placebo, Vyndaqel reduced the risk of all-cause death across all subgroups (wild-type, hereditary, NYHA functional classes I, II, III): the risk of death was reduced by 29% in the wild-type subgroup (HR=0.71, 95% CI: 0.474–1.052) and by 31% in the hereditary subgroup (HR=0.69, 95% CI: 0.408–1.167). (4) Across wild-type andHeredityIn the subtype subgroup, compared with placebo, Vyndaqel demonstrated a consistent reduction in the decline of patient functional capacity as assessed by the 6-Minute Walk Test (6MWT) and in all domains of patient quality of life as assessed by the Kansas City Cardiomyopathy Questionnaire (KCCQ). (5) In the study, Vyndaqel was well tolerated and exhibited a safety profile comparable to that of placebo.
The approval was also based on evaluation results from the 61 mg free acid form of tafamidis. The evaluation demonstrated that one 61 mg capsule of tafamidis free acid is equivalent to an 80 mg dose of tafamidis meglumine (four 20 mg capsules). The safety of the 61 mg dose was not evaluated in the ATTR-ACT trial. The 61 mg tafamidis capsule was developed to facilitate patient use and is suitable for once-daily administration.
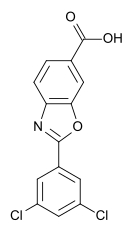
Molecular structure of tafamidis (from Wikipedia)
ATTR amyloidosis is a rare, progressive disease characterized by the abnormal accumulation of amyloid deposits composed of misfolded transthyretin protein in various organs and tissues throughout the body. Amyloidosis can affect many organs and systems, including the peripheral nervous system, as well as the heart, kidneys, gastrointestinal tract, and eyes. ATTR-CM and ATTR-PN are two manifestations of this disease.
ATTR-CM is a rare, fatal, and severeDiagnosisATTR-CM is a disease associated with progressive heart failure. ATTR-CM is caused by the instability of transthyretin (TTR), a transport protein composed of four identical subunits (tetramer). In ATTR-CM, when the unstable tetramer dissociates, it leads to heart failure, causing misfolded proteins to aggregate into amyloid fibrils and primarily deposit in the heart.
Tafamidis is an oral small-molecule drug that stabilizes TTR. In the United States and the European Union, tafamidis was granted orphan drug designation for the treatment of ATTR-CM in 2011. In 2017, the FDA also granted tafamidis Fast Track designation; in March 2018, Japan’s Ministry of Health, Labour and Welfare (MHLW) granted tafamidis Sakigake designation; in May 2018,FDATafamidis Granted Breakthrough Therapy Designation. (Bioon.com)
Original source: European Commissionapproves VYNDAQEL®, the First Treatment in the EU for Transthyretin Amyloid Cardiomyopathy (ATTR-CM)