Nature Medicine Publishes BangWise Scientists' Breakthrough: Next-Generation Base Editing Technology Holds Promise for Curing β-Thalassemia and Sickle Cell Disease

BRL Medicine
Cell and Gene Therapy Drug Developer
On March 17, 2020, the internationally renowned academic journal Nature Medicine published online the research findings titled “Therapeutic base editing of human hematopoietic stem cells” by Dr. Wu Yuxuan, a scientist at BRL Medicine, in the field of gene therapy.This study confirms that next-generation single-base editing technology holds promise for the complete cure of a series of genetic diseases caused by β-globin mutations [1].。
Jing Zeng from Professor Daniel Bauer’s laboratory at Harvard Medical School and Boston Children’s Hospital, Yuxuan Wu, a researcher at East China Normal University, and Chunyan Ren from Boston Children’s Hospital served as co-first authors. This marks another major breakthrough in the field of gene therapy for thalassemia by the scientific team at BRL Medicine, following their previous publications in journals such as Nature Medicine and Cell Research over the past year.

Thalassemia is the most widely distributed monogenic inherited disease globally, affecting the largest population. According to data from the "Blue Book on Prevention and Control of Thalassemia in China (2015)," there are currently approximately 30 million thalassemia gene carriers in China, involving a family population of 100 million. Additionally, there are about 300,000 patients with moderate to severe thalassemia, and this number is increasing at an annual rate of approximately 10%. Allogeneic hematopoietic stem cell transplantation is currently the only curative treatment for thalassemia; however, it is extremely costly and matching donors is highly challenging, making gene therapy a promising new treatment option.
β-Thalassemia (β-Thal, Thalassemia)β-thalassemia is a hereditary disorder caused by mutations in the β-globin subunit, leading to abnormal adult hemoglobin (HbA) in patients. It is the most widely distributed and prevalent monogenic genetic disease globally. In China, the detection rate of β-thalassemia is 0.67% (approximately 9.35 million people). There are roughly 300,000 severe cases of thalassemia, primarily concentrated in Guangdong, Guangxi, Guizhou, Hainan, and Yunnan provinces. Allogeneic hematopoietic stem cell transplantation is a curative treatment for thalassemia; however, due to its high cost and the extreme difficulty in finding matched donors, gene therapy has emerged as a promising new therapeutic option.
γ-globin is a fetal hemoglobin subunit with functions similar to those of β-globin. The HBG genes encoding this protein remain intact in patients with the aforementioned anemias, but their expression is silenced during adulthood. Previous research by the scientific team at BRL Medicine has demonstrated that reactivating fetal γ-globin expression through gene editing techniques to compensate for defective β-globin holds significant promise as a therapeutic strategy for alleviating or even curing thalassemia [2-3].
March 2019,Dr. Wu Yuxuan, Scientist at BRL MedicineA paper published in Nature Medicine revealed that targeted editing of the BCL11A erythroid enhancer using gene-editing technology can reactivate γ-globin expression, thereby substituting for defective β-globin and holding promise for a curative approach to such diseases [2]. On January 8, 2020, Dr. Li Dali, a scientist at BRL Medicine, published another study in the prestigious international academic journal Cell Research. By establishing a highly efficient gene-editing system in hematopoietic stem cells to mimic naturally occurring γ-globin promoter mutations found in certain populations, this approach can reactivate fetal hemoglobin (HbF), providing safe and effective therapeutic targets and methods for a series of genetic disorders caused by β-globin mutations [3].
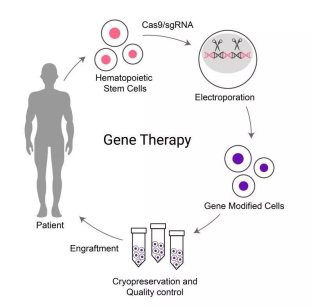
Figure 1. Strategies for treating thalassemia based on gene editing technology (from BRL Medicine's website)
Base Editors (BEs)Base editing refers to a gene-editing technology capable of inducing single-base changes in the genome. It enables precise conversion of cytosine (C) to thymine (T) or adenine (A) to guanine (G) at target gene loci without causing DNA double-strand breaks. In recent years, base editing has gradually become a focal point of research due to its potency, high efficiency, and ability to avoid DNA double-strand breaks, holding great promise for future gene therapy applications. Therefore, employing base editing to treat β-hemoglobinopathies (such as thalassemia) is highly likely to be an optimal strategy.
This study demonstrates that editing the BCL11A enhancer site in hematopoietic stem cells (HSCs) from patients with β-thalassemia and sickle cell anemia using an optimized base-editing technique, or directly repairing the mutated HBB gene, followed by autologous HSC transplantation, enables the in vivo differentiation of these cells into red blood cells producing functional hemoglobin. This approach holds the potential to completely cure these diseases. Furthermore, it confirms the significant potential of base-editing technology in gene therapy applications for HSCs, providing a novel therapeutic solution for the clinical treatment of thalassemia and sickle cell anemia.. The specific strategies are as follows:
First, this study achieved highly efficient gene editing in human CD34+ hematopoietic stem/progenitor cells (HSPCs) using an optimized single-base editing system. Subsequently, editing of the erythroid-specific enhancer at the BCL11A +58 locus resulted in downregulated BCL11A expression in erythroid cells. Upon in vitro erythroid differentiation of the edited CD34+ cells, BCL11A expression levels were significantly reduced, accompanied by a substantial increase in fetal hemoglobin (HbF) levels.
CD34+ cells derived from patients with the -28 type of thalassemia (the -28(A/G) mutation is one of the common mutation sites in the Chinese thalassemia population) were also edited. Furthermore, an attempt was made to simultaneously modify the dual sites HBB -28 and BCL11A +58 using a single base editor. The results showed that the differentiated red blood cells exhibited greater maturity, with their volume and morphology restored to levels nearly comparable to those of healthy cells.
Subsequently, the researchers confirmed that the edited stem cells maintained a remarkably high editing efficiency four months after transplantation into mouse bone marrow. Furthermore, hematopoietic stem cells (HSCs) exhibited a preference for C>T edits and were able to activate fetal hemoglobin expression in erythrocytes in vivo.
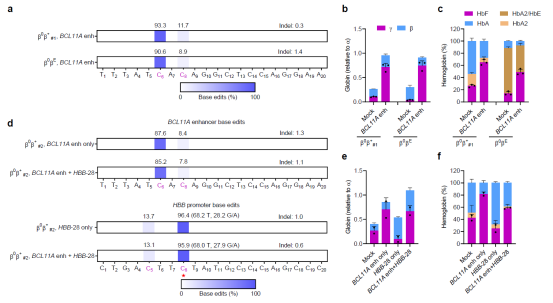
Figure 2. Editing hematopoietic stem cells from patients with β-thalassemia using single-base editing technology
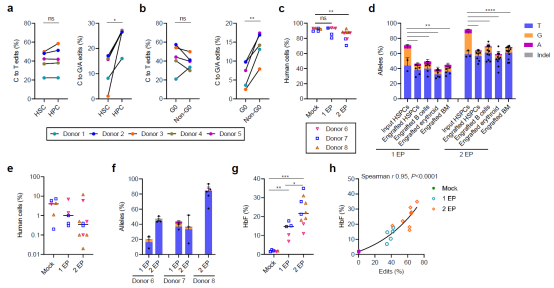
Figure 3. CD34+ cells edited with next-generation single-base editing tools can engraft long-term in mouse bone marrow while maintaining high-efficiency C>T base editing
Finally, researchers demonstrated that CD34+ cells derived from patients with sickle cell anemia could be efficiently edited and, upon transplantation into mice, successfully reconstitute the human hematopoietic system. Meanwhile, the elevated levels of fetal hemoglobin (HbF) in erythrocytes were sufficient to restore normal cell morphology and resist sickling.
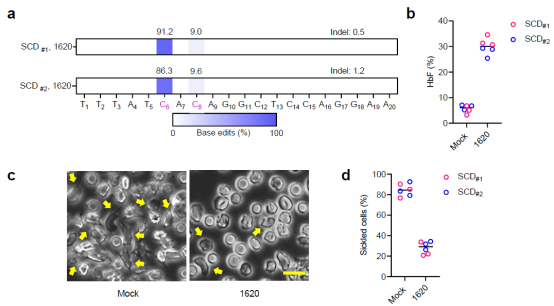
Figure 4. Erythrocytes derived from CD34+ cells of sickle cell anemia patients after single-base editing are resistant to sickling
According to statistics, the prevalence of β-thalassemia in China is 2.21%, with 48 identified variants. Gene therapy mediated by gene editing technology represents a major breakthrough in the field of precision medicine. The use of single-base editing technology to reactivate fetal hemoglobin expression also offers possibilities for personalized treatment of β-thalassemia.
BRL Medicine is actively advancing the translation of its scientific achievements into clinical applications. By collaborating with multiple top-tier medical institutions in China, the company aims to establish gene-editing-based gene therapy as a novel clinical treatment option for patients with β-thalassemia in China, with the potential to achieve a “one-time treatment, lifelong cure.” Compared to LentiGlobin (brand name: Zynteglo), a lentiviral therapy developed by Bluebird Bio—a leader in gene therapy—which is priced at a staggering $1.77 million and has become the second-most expensive drug globally, BRL Medicine’s therapeutic approach is more efficient, convenient, and safe, while significantly reducing costs. Furthermore, as there are currently no clinical trials in China utilizing this method for thalassemia treatment, BRL Medicine’s product is poised to become one of the first gene-editing therapies to enter clinical stages in the country.
References:
[1] Zeng J, Wu Y, Ren C, Bauer DE, et al. Therapeutic base editing of human hematopoietic stem cells. Nature Medicine, 2020
[2] Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarrotto CR, Clement MK, Cole MA, Luk K, Baricordi C, Shen AH, Esrick EB, Manis JP, Dorfman DM, Williams DA, Biffi A, Brugnara C, Biasco L, Brendel C, Pinello L, Tsai SQ, Wolfe SA, Bauer DE (2018) Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nature Medicine, 2019
[3] Wang L, Li L, Ma Y, Hu H, et al. Reactivation of γ-globin Expression through Cas9 or Base Editor to Treat β-Hemoglobinopathies. Cell Research, 2020,1