Roche's Risdiplam Demonstrates Significant Improvement in Survival and Motor Milestones in Infants with Type 1 Spinal Muscular Atrophy (SMA)
Apr 29, 2020 17:14
CST Updated
17:14

Roche
Oncology Drug Research, Development, and Manufacturing

April 29, 2020 News /Bio ValleyBIOON/ -- Roche recently announced one-year data from the pivotal global FIREFISH Part 2 study (NCT02913482) evaluating risdiplam for the treatment of infants with Type 1 spinal muscular atrophy (SMA). The results showed that the study met its primary endpoint:Risdiplam significantly improved survival rates and motor milestones in infants with type 1 SMA.These data were selected for presentation at the 72nd Annual Meeting of the American Academy of Neurology (AAN) and will be presented virtually in the coming weeks.ConferenceAvailable online.
Risdiplam is a splicing modifier of the survival motor neuron 2 (SMN2) gene, developed for the treatment of all types (Type 1, Type 2, and Type 3) of spinal muscular atrophy (SMA). Currently, the New Drug Application (NDA) for risdiplam is under review by the U.S. FDA. In early April this year,FDAExtend the target action date for this NDA by three months, to August 24. This extension of the review cycle is due to Roche’s submission of additional data, which helps ensure broad access to risdiplam treatment for patients with spinal muscular atrophy (SMA). These data include 12-month efficacy and safety results from the pivotal SUNFISH Part 2 study (n=180), the only placebo-controlled study conducted in patients aged 2–25 years with Type 2 or Type 3 SMA.
If approved,Risdiplam will become the first oral medication for the treatment of all three types of SMA.As part of the collaboration with the SMA Foundation and PTC Therapeutics, Roche led the clinical development program for risdiplam. If approved, Roche will be responsible for the commercialization of risdiplam in the United States.
FIREFISH is an open-label, multicenter Phase II/III study evaluating the safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and efficacy of risdiplam in infants aged 1 to 7 months with Type 1 spinal muscular atrophy (SMA). The study comprises two parts: an exploratory dose-finding part (Part 1, n=21) and a confirmatory part (Part 2, n=41). In Part 2, patients will receive oral risdiplam at the dose selected in Part 1 for 24 months. The primary endpoint is the proportion of infants able to sit unsupported for at least 5 seconds after 12 months of treatment, as assessed by the Motor Scale of the Bayley Scales of Infant and Toddler Development, Third Edition (BSID-III).
The one-year data from Part 2, released this time, demonstrate that the study met its primary endpoint: infants with Type 1 spinal muscular atrophy (SMA) treated with risdiplam showed statistically and clinically significant improvements in motor milestones. Specifically, 29% of infants (12/41, p<0.0001) were able to sit unsupported for at least 5 seconds at Month 12 of treatment. In the natural history of Type 1 SMA, no infants achieve this milestone. Furthermore, based on assessments using the Hammersmith Infant Neurological Examination-2 (HINE-2), 18 infants (43.9%) were able to hold their heads upright, 13 infants (31.7%) were able to roll sideways, and 2 infants (4.9%) were able to stand with support.
At the time of analysis, the median treatment duration was 15.2 months, and the median age was 20.7 months. Survival was observed in 93% (38/41) of infants, and 85.4% (35/41) remained event-free. In the natural history cohort, the median age at death or initiation of permanent ventilation was 13.5 months in the absence of treatment. Three infants experienced fatal complications within the first three months of treatment, all of which were considered by the investigators to be unrelated to risdiplam. The CHOP-INTEND score increased by at least 4 points in 90% (37/41) of infants, and 56% (23/41) achieved a score greater than 40; the median increase was 20 points. In contrast, CHOP-INTEND scores declined over time in untreated infants with type 1 SMA.
In an exploratory endpoint, among infants who survived to 12 months, 95% (36/38) retained swallowing ability and 89% (34/38) were able to feed orally. In contrast, in the natural history cohort, all infants with type 1 SMA older than 12 months required feeding support.In the FIREFISH study, the safety profile of risdiplam was consistent with previously reported data, and no new safety signals were identified.
Dr. Levi Garraway, Chief Medical Officer and Global Head of Product Development at Roche, stated: “These results confirm the clinical significance of risdiplam for infants with this advanced and refractory disease, including many whose disease had already progressed significantly prior to initiating treatment. We thank the SMA community for their collaboration, particularly the 62 families from around the world who participated in Part 1 and Part 2 of the FIREFISH study.”
Laurent Servais, Investigator of the FIREFISH study and Professor of Pediatric Neuromuscular Diseases at the MDUK Oxford Neuromuscular Centre, stated: “These results are particularly encouraging because the median age at enrollment was 5.3 months, meaning these infants already had progressive disease. Preserving swallowing function is especially important as it facilitates feeding in infants and indicates that risdiplam has a significant impact on bulbar function.”
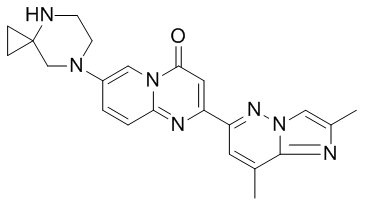
Risdiplam Chemical Structure (Image Source: medchemexpress.cn)
Risdiplam is an oral liquid, a survival motor neuron 2 (SMN2) splicing modifier designed to continuously increase and maintain SMN protein levels in the central nervous system and peripheral tissues. Growing clinical evidence demonstrates that spinal muscular atrophy (SMA) is a multisystem disease, and the loss of SMN protein may affect many tissues and cells beyond the central nervous system. Following oral administration, risdiplam exhibits systemic distribution, sustainably increasing SMN protein levels in both the central nervous system and peripheral tissues, and has been shown to improve motor function in patients with type 1, type 2, and type 3 SMA.
Risdiplam is poised to become the first oral medication for the treatment of all three types of spinal muscular atrophy (SMA). As an oral liquid formulation, risdiplam, if approved, would be the first therapy available for home administration to patients with SMA. Previously,FDARisdiplam has been granted orphan drug designation and fast track designation.
Risdiplam is being investigated in a comprehensive clinical trial program for spinal muscular atrophy (SMA), enrolling patients ranging from newborns to adults aged 60 years, including presymptomatic individuals and those who have previously received other SMA therapies. ThisClinical TrialThe project aims to reach a broad, real-world population of patients with this disease, ensuring that all eligible patients have access to risdiplam treatment.
As part of its ongoing commitment to patients with spinal muscular atrophy (SMA), Roche is also in Brazil, Chile, Indonesia, Russia, South Korea,Registration in Mainland China is imminent, the company is currently on track to submit marketing authorization applications to the European Medicines Agency (EMA) and other international markets in mid-2020.
Spinraza: The World’s First SMA Treatment Drug, Approved in China
Spinal Muscular Atrophy (SMA) is a motor neuron disease that causes muscle weakness and atrophy. It is an autosomal recessive genetic disorder caused by gene defects, affecting muscles throughout the patient's body. Patients primarily present with generalized muscle atrophy and weakness, progressively losing various motor functions, including breathing and swallowing. SMA is the leading cause of death in infants under the age of two.Heredity"Killer disease," this condition is a relatively common "rare disease," with an incidence rate of 1:6000-1:10000 in newborns. According to relevant reports, there are currently approximately 30,000 to 50,000 SMA patients in China.
In December 2016, Spinraza (nusinersen), a drug developed by Biogen and its partner Ionis, was approved, becoming the first therapy worldwide for spinal muscular atrophy (SMA). This medication is an antisense oligonucleotide (ASO) administered via intrathecal injection, delivering the drug directly into the cerebrospinal fluid (CSF) surrounding the spinal cord. It modifies the splicing of SMN2 precursor messenger RNA (pre-mRNA), thereby increasing the production of full-length functional SMN protein. In patients with SMA, insufficient levels of SMN protein lead to the degeneration of motor neuron function in the spinal cord. Clinical studies have demonstrated that treatment with Spinraza significantly improves motor function in patients with SMA.
In May 2019, fromNovartisThe gene therapy Zolgensma (onasemnogene abeparvovec) was approved, becoming the world’s first gene therapy for the treatment of spinal muscular atrophy (SMA). Administered as a single, one-time intravenous infusion, the drug enables sustained expression of the SMN protein to halt disease progression, addresses the root cause of SMA, and holds promise for long-term improvement in patients’ quality of life.
In the Chinese market, Spinraza was approved in late February 2019 for the treatment of patients with 5q spinal muscular atrophy (5q-SMA). This approval made Spinraza the first drug for treating SMA in the Chinese market. 5q-SMA is the most common type of SMA, accounting for approximately 95% of all SMA cases. This type of SMA is caused by mutations in the SMN1 (survival motor neuron 1) gene on chromosome 5, hence the name 5q-SMA. (Bioon.com)
Original Source: Roche’s risdiplam shows significant improvement in survival and motor milestones in infants with Type 1 spinal muscular atrophy (SMA)