Genentech's Bispecific Antibody Platform Demonstrates Significant Reduction in On-Target, Off-Tumor Toxicity Through Tumor-Specific Anchor Receptors

Genentech
Pharmaceutical R&D Manufacturer


Genentech’s work proposes a straightforward engineering logic: if toxicity arises from specific cell types, identify an “anchor receptor” expressed on tumor cells but absent on normal cells, and pair it with a low-affinity inhibitory arm. The bispecific antibody (bsAb) restores inhibitory activity through avidity only when it simultaneously binds both the anchor receptor and the target receptor. This “AND logic gate” molecular design shifts cellular selectivity from the target receptor itself to the co-expression pattern of the target and anchor receptors.

Wnt/FZD Inhibition Is Effective, but Intestinal Toxicity Is a Hard Constraint
The study initially focused on the RNF43-mutant subgroup in pancreatic ductal adenocarcinoma (PDAC). RNF43 is an E3 ubiquitin ligase, and its mutations lead to the accumulation of Frizzled (FZD) receptors and sustained activation of the Wnt pathway. Large-scale functional screening data from DepMap indicate that Wnt-related genes such as CTNNB1 and TCF7L2 exhibit strong lineage-selective dependency in colorectal adenocarcinoma (COAD) and PDAC, a feature not observed in other tumor types.
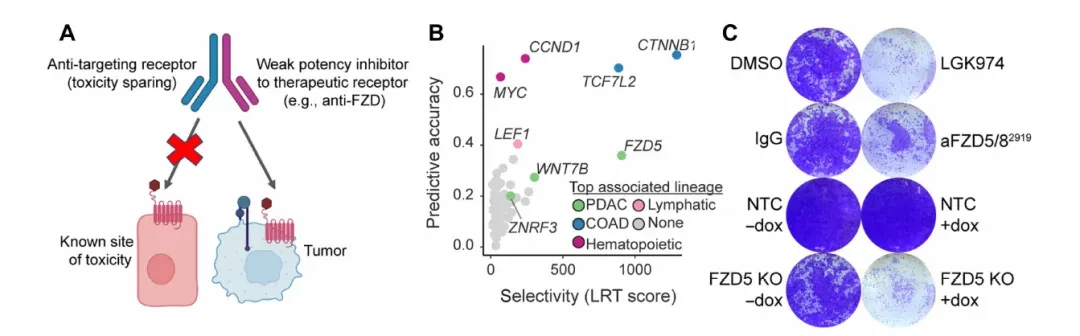
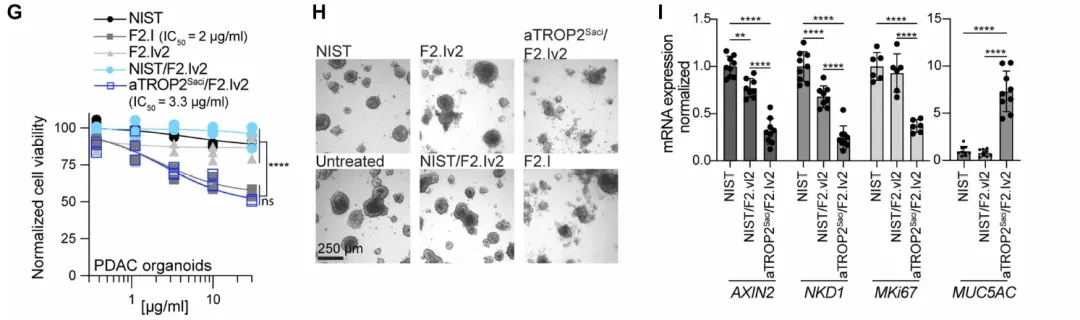
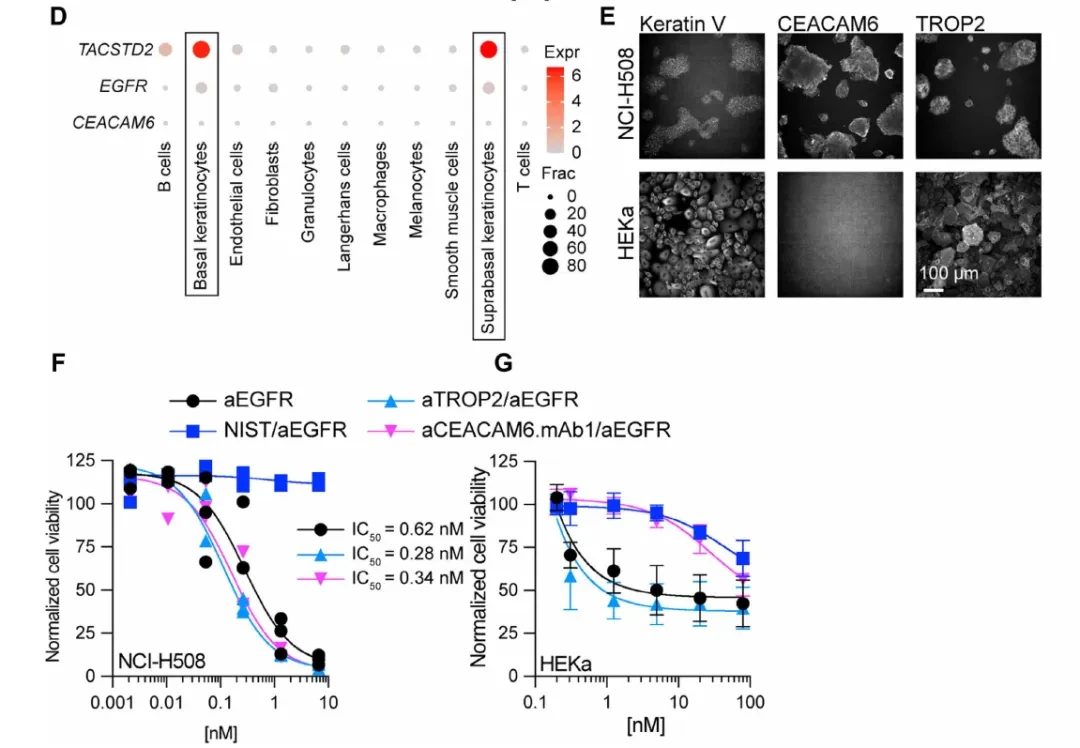
To validate FZD5 dependence, researchers established a doxycycline-inducible FZD5 knockout system in HPAF-II cells (harboring the RNF43 p.E174Ter mutation) and evaluated two FZD-inhibitory antibodies: aFZD5/82919, which targets only FZD5/8, and F2.I, a pan-FZD (1/2/4/5/7/8) inhibitory antibody. Colony formation assays demonstrated that FZD5 knockout alone was sufficient to block HPAF-II cell growth; mRNA levels of the Wnt target genes AXIN2 and NKD1 were significantly reduced following knockout, while the differentiation marker MUC5AC was correspondingly upregulated (Fig. 1C-D). In multiple PDAC cell lines, the growth-inhibitory activity of F2.I (pan-FZD) was generally stronger than that of aFZD5/82919, with an IC50 < 1 nM, suggesting that inhibition of FZD5 alone is insufficient to completely block the pathway.
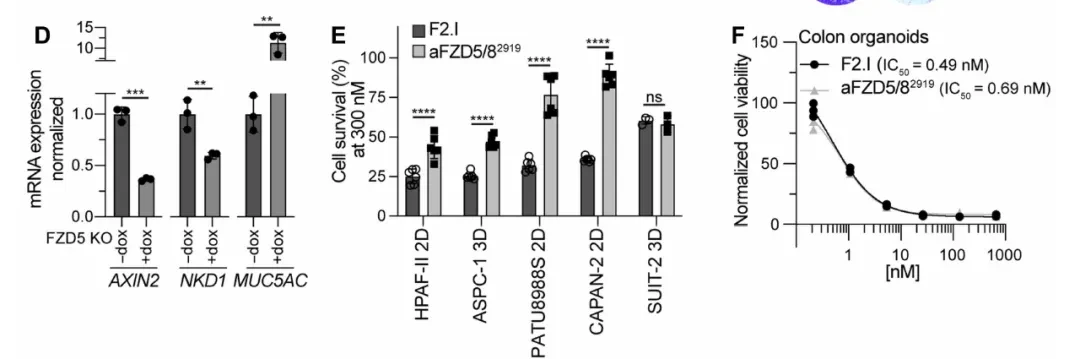
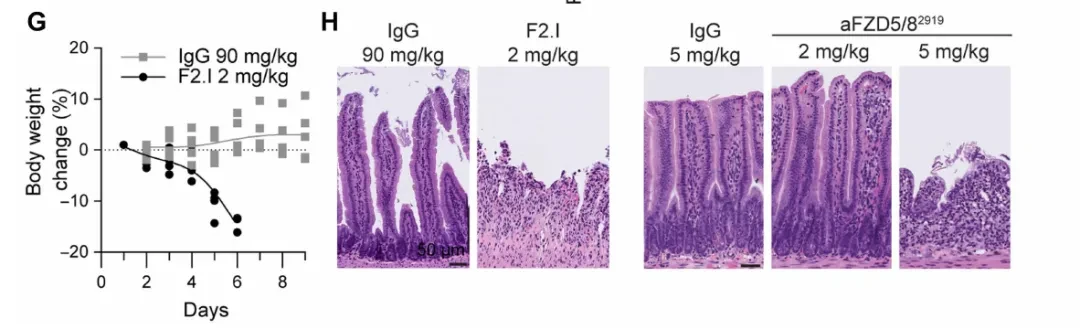
However, when researchers administered F2.I (2 mg/kg) to C57BL/6 mice, the animals rapidly exhibited weight loss and severe intestinal pathology, including villous atrophy and shedding (Fig. 1G-H). Normal colon organoids also showed potent growth inhibition in response to F2.I, with an IC50 < 1 nM. Together, these two sets of data point to the same conclusion: the FZD/Wnt pathway is indispensable for the maintenance of intestinal stem cells (the LGR5+ subset), and thus cannot be targeted via systemic administration of traditional single-target antibodies.
scRNA-seq Screening for Tumor-Specific Anchor Receptors
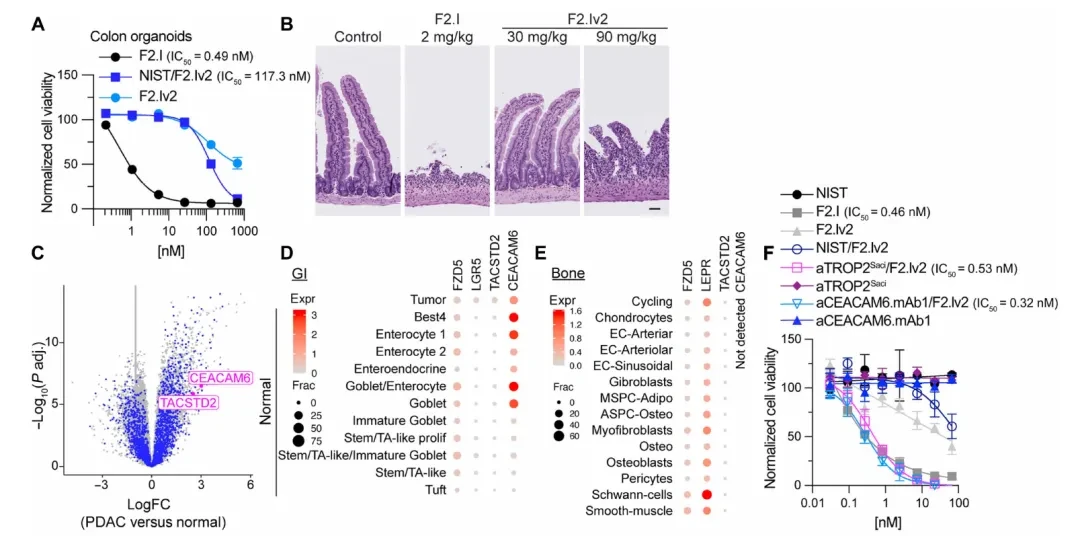
Once the source of toxicity is identified, the question becomes: which receptor is highly expressed on PDAC tumor cells but absent or expressed at very low levels in key stem cell populations in the intestine and bone?
The researchers first analyzed bulk RNA-seq datasets from 36 human PDAC tumors and paired normal pancreatic tissues to obtain an initial list of highly expressed receptors (Figure C). Subsequently, human colon (The following figureD) and bone marrow (The figure belowE) The single-cell RNA sequencing (scRNA-seq) datasets were filtered individually, using LGR5 (a marker for intestinal stem cells) and LEPR (a marker for skeletal stem cells) as surrogate markers for toxic cells, to overlay and screen the expression distribution of candidate targets.
TACSTD2 (encoding TROP2) and CEACAM6 (carcinoembryonic antigen-related cell adhesion molecule 6) stood out in this screening: both are highly expressed in PDAC tumor cells, exhibit negligible expression in LGR5+ intestinal stem cells, and show minimal to no expression in LEPR+ bone marrow stem cells.
Based on this, the researchers generated two bsAbs: aTROP2Saci/F2.Iv2 (TROP2 anchor + attenuated pan-FZD arm) and aCEACAM6.mAb1/F2.Iv2 (CEACAM6 anchor + attenuated pan-FZD arm). F2.Iv2 is an attenuated version of F2.I, with affinities >50 nM for all FZD subtypes and no growth inhibitory activity when used alone. However, both bsAbs exhibited activity comparable to or even superior to that of the parental F2.I in multiple PDAC cell lines, including HPAF-II, CAPAN-2, and ASPC-1 (IC50 < 1 nM,The figure belowF). The critical control group—ASPC-1 cells lacking TROP2 expression—showed no response to aTROP2Saci/F2.Iv2; inhibitory activity was restored upon doxycycline-induced TROP2 overexpression.
Patient-derived PDAC organoids (The figure belowIn panels G–H, F2.I and the target-engaging bsAb exhibited similar growth inhibitory potency, whereas human colon organoids (which do not express TROP2 and CEACAM6) were sensitive only to F2.I and showed no response to the target-engaging bsAb. This precise receptor dependency recapitulates in vivo selectivity at the tissue level.
qPCR Quantification (The figure belowI) Further confirmation: After 48 hours of treatment with the targeted bsAb, mRNA levels of AXIN2, NKD1, and MKi67 in HPAF-II cells were significantly decreased, while MUC5AC levels were correspondingly increased. The effect was consistent with that of the parental F2.I, confirming that the activity was due to specific inhibition of the Wnt pathway.
Dual Validation of In Vivo Efficacy and Gastrointestinal Tolerance
After meeting the in vitro criteria, the researchers proceeded to the HPAF-II subcutaneous xenograft model. In the pharmacodynamic study (Figure A below), administration of aTROP2Saci/F2.Iv2 at 30 mg/kg for 48 hours significantly decreased mRNA levels of AXIN2, NKD1, and LGR5 in tumor tissues, while increasing MUC5AC levels; in contrast, the non-targeting F2.Iv2 showed only minimal effects.
Efficacy Study (Figure belowB) The results showed that aTROP2Saci/F2.Iv2 at doses of 5 mg/kg and 30 mg/kg both significantly reduced tumor volume, with no significant changes in body weight. The bivalent F2.Iv2 (without targeting arm) at 30 mg/kg exhibited weaker efficacy and caused initial body weight loss, leading to forced discontinuation of treatment. The control groups (non-targeting F2.Iv2 or isotype control) showed no significant antitumor activity.
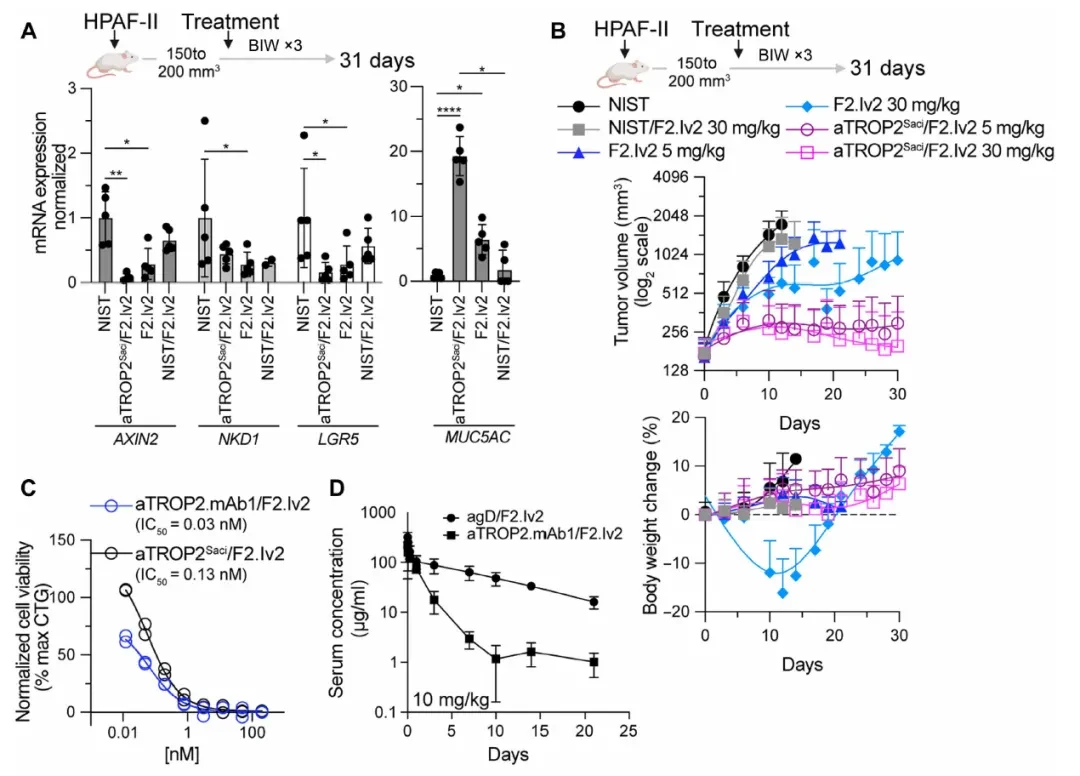
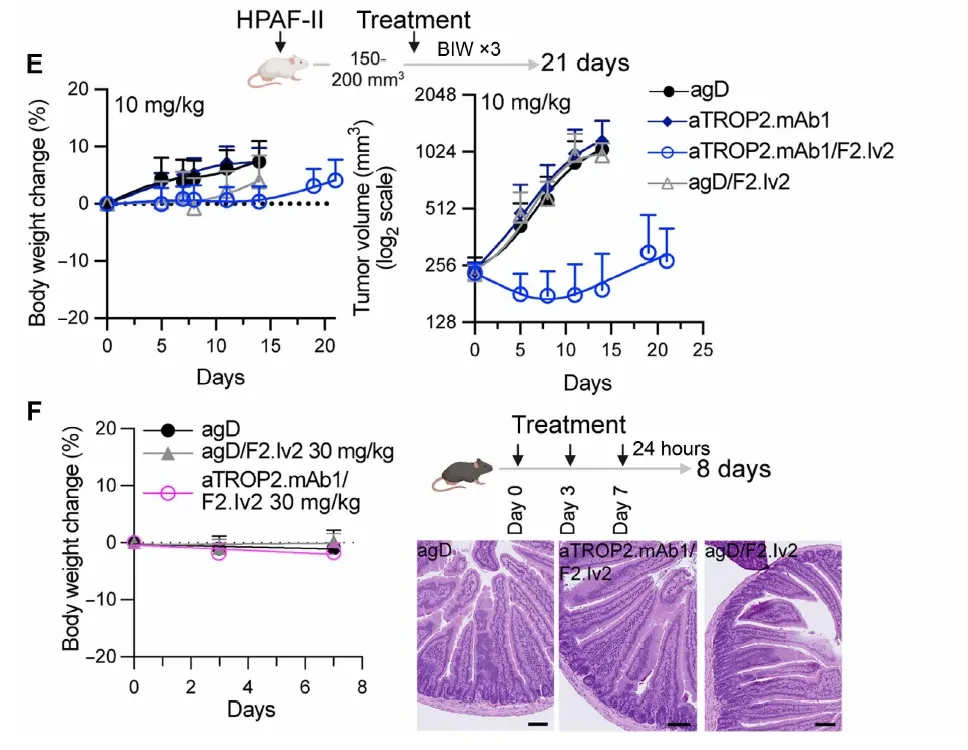
Given that aTROP2Saci does not bind to mouse TROP2, researchers further screened for cross-reactive antibodies by immunizing with the TROP2 protein, yielding the antibody aTROP2.mAb1 (with affinities of 0.5 nM for human TROP2 and 1.2 nM for mouse TROP2), and generated a new bispecific antibody, aTROP2.mAb1/F2.Iv2. Pharmacokinetic studies (aboveFigureD) The data show that the bsAb was cleared slightly faster in healthy mice than in the control group (suggesting some degree of TROP2 tissue distribution), but maintained concentrations >1 μg/mL (6.7 nM), approximately 200-fold higher than the in vitro IC50. Administration at 10 mg/kg demonstrated potent antitumor activity against HPAF-II xenografts, with minimal changes in body weight (Figure belowE)。
Key Intestinal Tolerance Verification (The figure belowF): In immunocompetent, non-tumor-bearing C57BL/6 mice, three administrations of aTROP2.mAb1/F2.Iv2 at 30 mg/kg (within 8 days) resulted in no changes in body weight and no significant abnormalities in small intestine histopathology, whereas a single dose of F2.I at 2 mg/kg caused severe intestinal pathology. The comparison between the two groups validated the core hypothesis of the molecular design: that targeting anchor receptor-driven activity selectivity holds true in vivo as well.
Mechanism of Activation of Inert FZD Antibodies
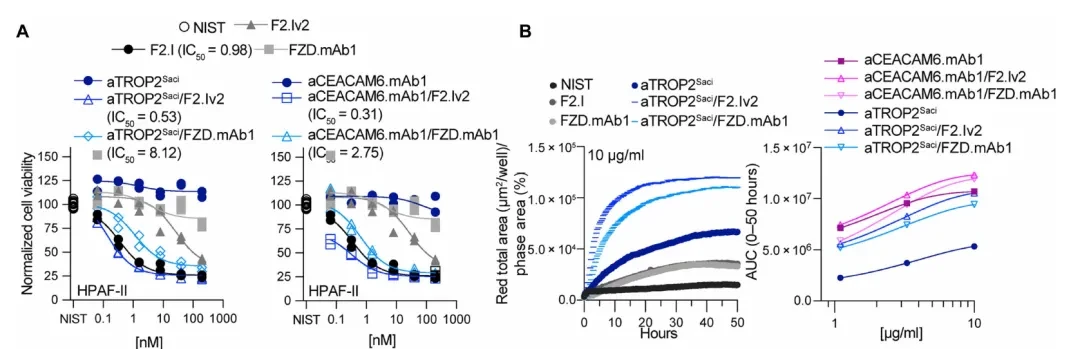
One phenomenon in the above results warrants separate explanation: even when F2.Iv2 was replaced with the novel antibody FZD.mAb1, which itself exhibits virtually no inhibitory activity (with affinities of 15–33 nM for FZD1/2/5/7/8 and no growth inhibition against HPAF-II when used alone), pairing it with TROP2 or CEACAM6 still achieved growth inhibition comparable to that of the F2.Iv2-based bispecific antibody (bsAb) (less than a 10-fold difference; see Figure A below).
This excludes the possibility of simple "affinity recovery" as the sole mechanism, suggesting the presence of additional mechanisms.
Internalization Assay (Panel B below): The amount of internalization was assessed using pH-sensitive fluorescein-labeled Fab fragments. The internalization rates of both bispecific antibodies (bsAbs), aTROP2Saci/F2.Iv2 and aTROP2Saci/FZD.mAb1, were significantly faster than that of the aTROP2Saci monoclonal antibody alone; whereas FZD.mAb1 or F2.Iv2 alone showed negligible internalization. A similar pattern was observed for CEACAM6-targeting bsAbs.
Mechanism of Action Inference: Upon binding to the anchor receptor, the bispecific antibody (bsAb) is rapidly internalized into low-pH endosomes, concurrently carrying the bound FZD receptor into the cell. This process blocks Wnt ligand binding through spatial segregation of the receptor, a mechanism that neither requires sustained high-affinity FZD inhibition nor extensive receptor degradation.
Transcriptomic analysis (RNA-seq, Figure D) further confirmed that the global gene expression changes induced by aTROP2Saci/FZD.mAb1 and F2.I in HPAF-II cells were highly correlated (r=0.767), with the Wnt gene set (Figure F) showing consistent downregulation. Treatment with FZD.mAb1 alone had no effect on the Wnt gene set, whereas the bispecific antibody format fully recapitulated the inhibitory effects on the FZD pathway.
Strategy Extension: EGFR and FGFR1-Targeting Bispecific Antibody
Encouraged by the results of FZD, the researchers extended the same design logic to two other targets with well-defined toxicity limits.
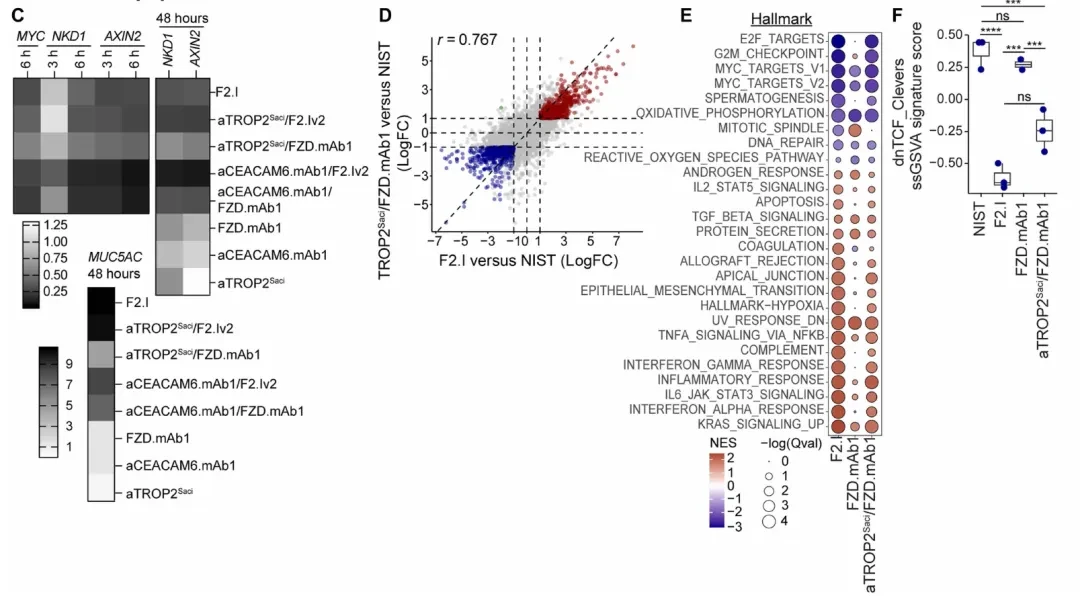
FGFR1/TROP2: The nephrotoxicity of FGFR1 inhibitors stems from FGF23-FGFR1/Klothoβ signaling in proximal renal tubular cells. scRNA-seq data (Figure A below) show that FGFR1 and Klothoβ are co-expressed in renal proximal tubule cells, whereas TACSTD2 (TROP2) is primarily expressed in renal collecting ducts, indicating distinctly different cell types.
Based on this, the TROP2 anchor was paired with the FGFR1 inhibitory arm (IMC-H7) to generate a bispecific antibody (bsAb). Western blot analysis (Figure B below) demonstrated that following FGF1 activation, either the aTROP2/aFGFR1 bsAb or the small-molecule FGFR1–3 inhibitor AZD4547 completely blocked pFGFR1 and pPLCγ phosphorylation in CAL120 TROP2+ breast cancer cells; whereas the same bsAb exhibited no inhibitory effect on FGF1/FGFR1 signaling in G-402 renal cells. Cell proliferation assays (Figure C below) further confirmed that blockade of the FGFR1 pathway translated into effective growth inhibition in breast cancer cells (TROP2+); moreover, the monovalent FGFR1-binding format eliminated the receptor agonistic effect observed with the bivalent FGFR1 IgG.
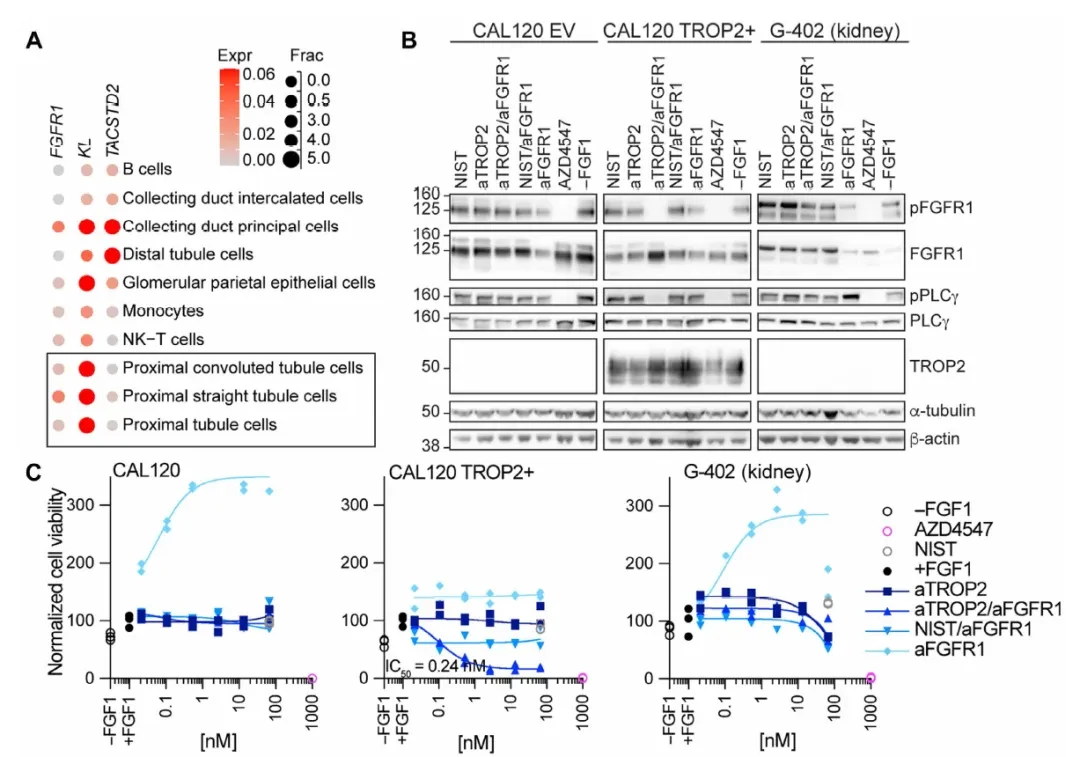
EGFR/CEACAM6: The cutaneous toxicity of EGFR inhibitors stems from the dependence of keratinocytes on EGFR signaling. Single-cell skin data (Figure D below) show that CEACAM6 is expressed at very low levels in cutaneous keratinocytes, whereas TACSTD2 (TROP2) is highly expressed. Therefore, it is more rational to use CEACAM6 as the anchor receptor.
Immunofluorescence (Figure E) confirmed the co-expression of CEACAM6 and TROP2 in the CRC cell line NCI-H508, whereas only TROP2 was detected, with no CEACAM6, in adult human keratinocytes (HEKa). In functional assays (Figures F–G), the aCEACAM6/aEGFR bispecific antibody (bsAb) exhibited growth inhibitory activity against NCI-H508 (a cetuximab-sensitive CRC cell line) comparable to that of the bivalent aEGFR IgG. In contrast, the bsAb showed only slight effects on HEKa keratinocytes at concentrations more than 100-fold higher. Conversely, both aEGFR IgG and the aTROP2/aEGFR bsAb (which uses TROP2 as an anchor, despite its high expression in HEKa) demonstrated potent toxicity against HEKa cells.This >100-fold therapeutic window gap stands in sharp contrast to the activity of the TROP2 anchor.
GenentechThis work distills a set of reproducible workflows for the design of targeted antagonists:
1)Identify Cytotoxic Cells: Pinpoint the specific cell subpopulations driving toxicity (intestinal LGR5+ stem cells, renal proximal tubule cells, and skin keratinocytes) via scRNA-seq, rather than relying on the coarse resolution of bulk RNA data.
2)Screening for Tumor-Specific Anchor Receptors: Select anchors from the intersection of high tumor expression and low/absent toxic cell expression. TROP2 and CEACAM6 recur across three target scenarios, suggesting their broad applicability in multiple tumor types (PDAC, breast cancer, CRC).
3)Weakened Inhibitory Arm:High-affinity inhibitory activity is not required as a standalone property; instead, target inhibition is restored through the synergy of avidity and internalization in the bispecific antibody (bsAb) format, while maintaining minimal activity on normal cells lacking the anchor receptor.
4) Receptor Ratio Is a Key Parameter: A ratio of anchor receptors to target receptors of at least 5:1 is required to support sufficient avidity binding—this provides a quantifiable structural constraint for the screening of candidate molecules.
Exploring new safety boundaries in targeted drug development beyond ADCs and TCBs is a worthwhile direction for research.


Warm Reminder
