Accelerated Rational PROTAC Design via AI and Molecular Simulations: A Dual-Driven Approach to Targeting 'Undruggable' Proteins

Galixir
AI Technology Empowers Drug Developers

Since the first proof-of-concept of Proteolysis Targeting Chimeras (PROTACs) in 2001, PROTACs have become a revolutionary tool for the selective degradation of target proteins through the ubiquitin-proteasome system. PROTACs consist of three parts: one targeting the protein of interest(POI)Ligand(Also known as warhead)A ligand that recruits E3 ubiquitin ligase, and a chemical linker that connects the two ligands. Due to this bifunctional structure, PROTACs have the ability to simultaneously bind to the target protein and E3 ubiquitin ligase, forming an active ternary complex. Therefore, PROTACs only need to briefly bind to the target protein to induce ubiquitination and degradation. In addition, the fact that PROTACs do not need to occupy a druggable active site makes it possible to use all surface binding sites of the target protein to modulate "undruggable" targets. However, the current design and optimization of PROTACs still require empirical iterative refinement, which presents certain limitations in the development strategy.
In the development of PROTACs, the most challenging issue is how to select an appropriate linker to form a suitable PROTAC-active ternary complex that exhibits degradation activity and target selectivity. Due to the complexity and dynamic nature of the ternary structure, designing the linker often presents a significant challenge. The length, composition, flexibility, and attachment sites of the linker can all have a substantial impact on the outcome. Additionally, another design challenge arises from the fact that PROTAC molecules often do not meet the common properties required for oral drugs. As multi-component molecules, their relatively large molecular weight leads to issues such as poor solubility, low permeability, poor bioavailability, and unpredictable Hook effects compared to traditional small molecules, which hinder the clinical translation of PROTACs. Therefore, rationally optimizing PROTAC molecules to overcome these problems under limited conditions remains a major challenge in this field.
Dual-Driven by Intelligence and Simulation Computing
PROTAC-RL Discovers Novel Lead Compound in Just 49 Days
To address this issue, the research team proposed a PROTAC rational design algorithm based on deep generative models—PROTAC-RL. The model takes a pair of E3 ligands and warheads as input, outputs the designed linkers, and utilizes reinforcement learning.(Reinforcement Learning,RL)Under the guidance, PROTAC molecules with specific properties are generated. Specifically, the research team first pre-trained a linker generation model using a Transformer neural network. Then, during the model training process, to overcome the issue of limited PROTAC training data, the model was pre-trained with a large number of quasi-PROTAC molecules that share a similar chemical space with PROTACs, followed by fine-tuning the model with real PROTACs and augmented data. Subsequently, the trained model was integrated into a memory-based reinforcement learning module with empirical reward functions to generate PROTACs with better pharmacokinetic properties.
As a proof of concept, the research team selected BRD4 as the target protein and generated more than 5,000 PROTACs. Leveraging supercomputing power, the team used high-throughput machine learning scoring functions and molecular dynamics simulations to cluster and screen these virtual molecules. Based on synthetic accessibility, the researchers ultimately selected, synthesized, and experimentally tested six PROTACs, three of which demonstrated inhibitory activity against BRD4. One lead compound also showed high anti-proliferative potency in tumor cell lines and exhibited favorable pharmacokinetics in mice.(Figure 1)Based on Galixir's long-term exploration and沉淀 in the PROTAC research direction, the large-scale parallel molecular dynamics simulation methods from the National Supercomputing Center in Guangzhou at Sun Yat-sen University, and the massive GPU computing power used for deep learning model training, the entire research process took only 49 days. This demonstrates that the combination of supercomputing, deep learning, and molecular dynamics can facilitate efficient and rational PROTAC design and optimization.
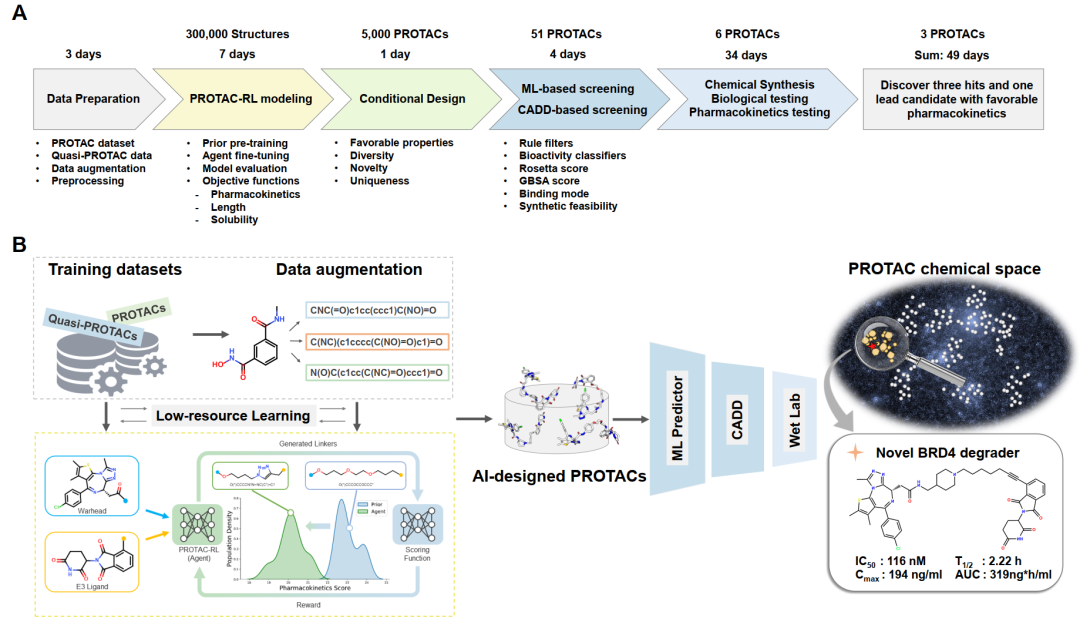
The PROTAC-RL model consists of two parts: the base generation model Proformer and the drug-likeness reinforcement learning model RL. In evaluating Proformer, researchers first split the PROTAC dataset in an 8:1:1 ratio for training, validation, and testing. For each test pair of warheads and E3 ligands, 10 candidate PROTACs were generated, and the percentage of reproducing the true PROTACs in the test set was used as the evaluation metric.(Reproducibility Rate)To evaluate performance, the researchers compared Proformer with other state-of-the-art fragment linking methods, including the graph learning-based method Delinker, the sequence-based method Syntalinker, and their retrained versions on the PROTAC training set. As shown in Figure 2-A, Proformer achieved a reproducibility rate of 43.0%, significantly outperforming the best existing baseline methods. The molecules generated by Proformer are also closer to the real chemical space of PROTACs compared to other methods.(Figure 2-B). At the same time, the ablation experiment(Figure 2-C)This once again proves the rationality of the model design. After being combined with reinforcement learning, the molecular scores generated by the PROTAC-RL model are also much higher than those of other model variants.(Figure 2-D)In a selected case, the PROTAC-RL model can generate specific linker properties based on different target score settings.(Fig. 2-E, F)Overall, compared with other methods, the PROTAC-RL model shows superior performance in terms of reproducibility, effectiveness, uniqueness, and novelty.
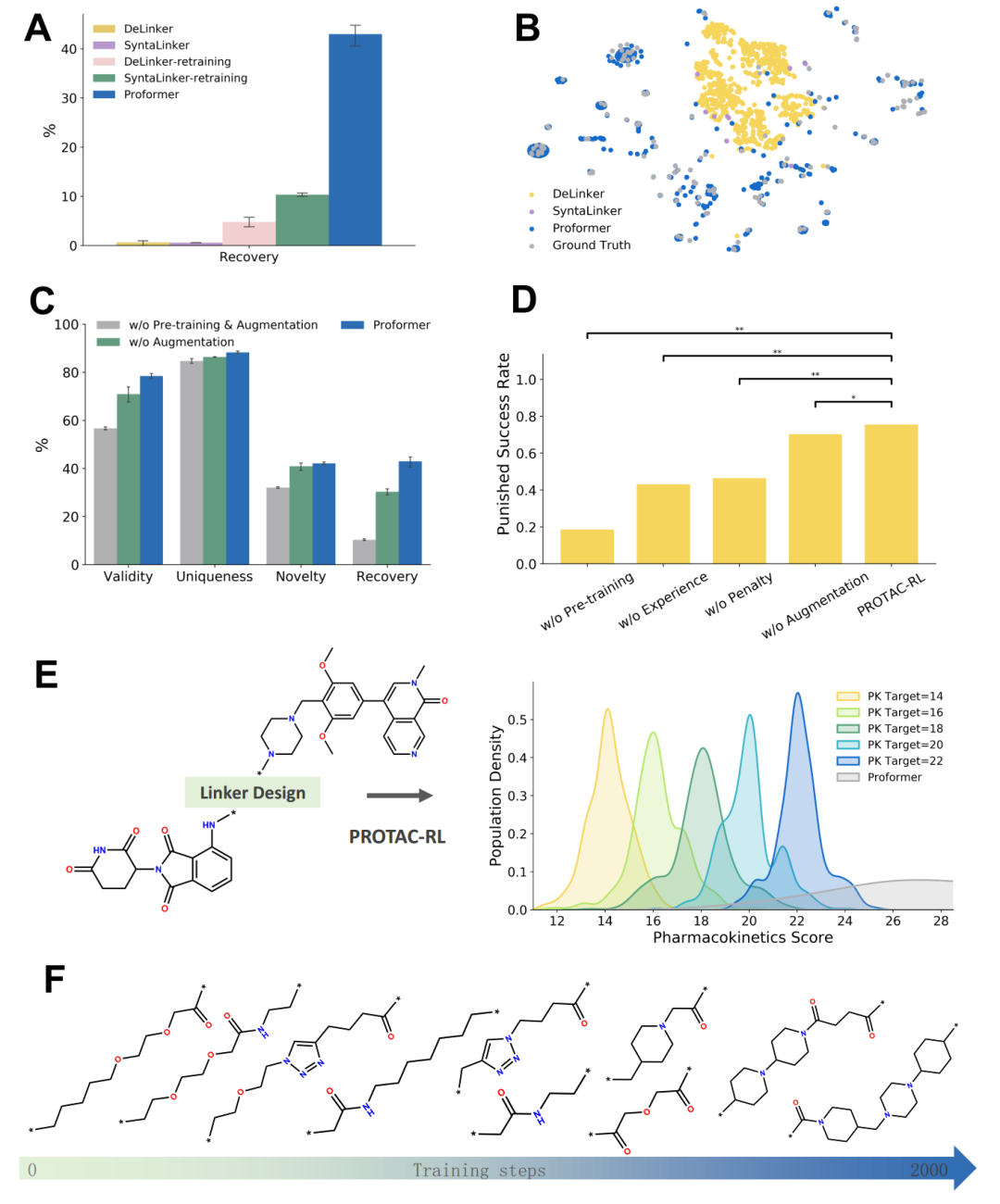
Figure 2. Comparison results of PROTAC-RL with the latest prediction methods and the results of ablation experiments
Validation Case: PROTAC Design Targeting BRD4 and Wet Lab Validation
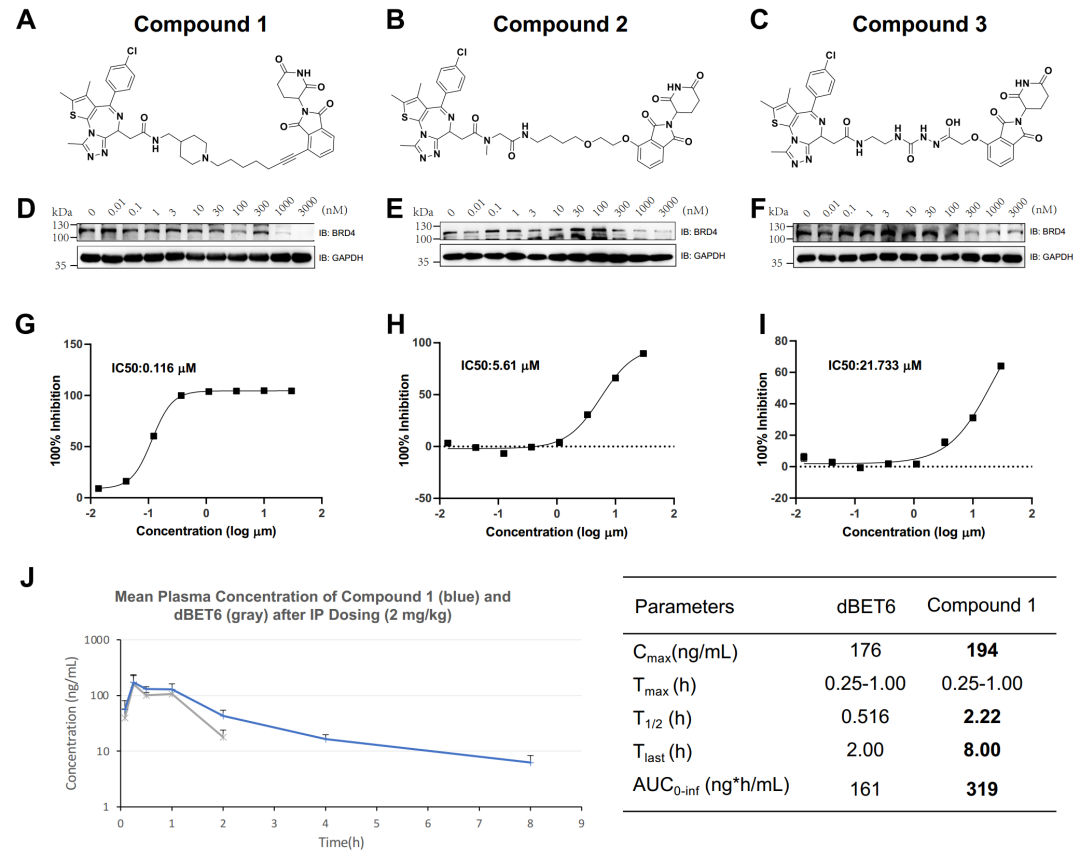
This research report outlines a fully automated computational framework that integrates reinforcement learning-driven deep generative models, machine learning, and molecular dynamics simulations for the rational design and optimization of PROTACs. In a case study targeting BRD4, Galixir's research team, in collaboration with Professor Yuedong Yang from the National Supercomputer Center in Guangzhou at Sun Yat-sen University, relied on "Tianhe-2" and combined cross-disciplinary expertise in supercomputing, AI, and biopharmaceuticals. By leveraging reinforcement learning-based molecular generation and physics-based molecular simulation techniques, the team utilized dual-driven intelligent and simulation computing to accelerate PROTAC drug discovery. Within just 49 days, they identified novel lead compounds with high degradation activity and excellent pharmacokinetic properties and completed wet-lab validation. This further demonstrates that integrating supercomputing, AI-driven computational strategies, and experimental approaches is a crucial method for obtaining effective drug candidates.