ARTHEx Biotech Secures $60M Series B Financing to Advance ATX-01, a Dual-Mechanism miRNA-Targeting Oligonucleotide Therapy for Myotonic Dystrophy Type 1

ARTHEx Biotech
Innovative Drug Developer
Myotonic Dystrophy Type 1 (DM1) is an autosomal dominant genetic disorder characterized primarily by myotonia and progressive muscle weakness, with high disability rates. After the onset of DM1, patients often exhibit multi-system damage, including cataracts, arrhythmias, insulin resistance, gastrointestinal dysfunction, gonadal abnormalities, and cognitive impairment. Severe cases may lose the ability to work and live independently due to late-stage complications, with secondary pulmonary infections and nutritional disorders potentially leading to death.
According to data released by the Myotonic Dystrophy Foundation in the United States, DM1 is estimated to affect 1 in 3,000 to 8,000 individuals worldwide.There is currently no specific treatment regimen.。
ARTHEx Biotech is a Spanish biotechnology company developing microRNA (miRNA) modulating drugs. miRNA is a class of non-coding RNA molecules that play a role in post-transcriptional gene expression regulation by binding to target mRNAs and inhibiting protein synthesis.
The company has recently completed a 42 million euro Series B financing. This funding will support ARTHEx Biotech to initiate clinical development of its DM1 treatment.
This round of investment was led by Columbus Venture Partners, SGEIC, and SAU Partners. New investors include the European Innovation Council (EIC) Fund, Hadean Ventures, and Sound Bioventures, along with existing investors Invivo Capital, AdBio Partners, and the Centre for the Development of Industrial Technology (CDTI).
Traditional beliefs hold that RNA molecules play a passive role in transmitting information for human protein synthesis, acting as messengers between DNA and proteins—namely, messenger RNA. However, with the advancement of research, more functions of RNA have been uncovered. For instance, RNA can help activate or deactivate genes, facilitate chemical reactions, cleave other RNA molecules, and transport amino acids to build proteins.
These functions of RNA have been further explored by biophysical chemists, leading to the development of RNA-based drugs. These RNA-targeted therapies are often used to develop treatments for rare genetic diseases that currently have no therapeutic options. For example, Spinraza (nusinersen), an injection used to treat spinal muscular atrophy (SMA) in both children and adults. It was approved by the FDA in 2016 and launched in China in February 2019 through a priority review process.
Spinraza is an antisense oligonucleotide. Oligonucleotides are typically short chains of fewer than 20 ribonucleotides that pair with mRNA or pre-mRNA through Watson-Crick base complementarity to achieve very high selectivity and precisely inhibit certain genes, silencing abnormally coded genes to regulate protein expression. For example, Viltolarsen, a treatment for Duchenne muscular dystrophy (DMD), is an antisense oligonucleotide therapy that promotes the production of functional dystrophin by masking exon 53 of the dystrophin gene.
There are many types of RNA-based oligonucleotide therapies under research, including antisense oligonucleotides (ASO), small interfering RNA (siRNA), and microRNA (miRNA). The range of treatable indications continues to expand, from the antisense oligonucleotide drug Viltolarsen for treating Duchenne Muscular Dystrophy (DMD), to the siRNA drug Leqvio for treating adult primary hypercholesterolemia.
However, there are some obstacles to the widespread application of oligonucleotide therapies. The main obstacle lies in the difficulty of effectively delivering oligonucleotides to target organs and tissues outside the liver. At the same time, off-target interference, sequence, chemical dependency, and toxicity also need to be carefully considered. In addition, due to the high R&D costs and long R&D cycles, such drugs often have a price ceiling — Nusinersen injection was once the famous "700,000-yuan-a-shot" expensive drug. But in the 2021 medical insurance negotiation, it was included in the medical insurance at a price lower than 33,000 yuan per shot.
DM1 is an autosomal dominant genetic disorder with the causative gene located at 19q13.3, caused by the abnormal expansion of a CTG trinucleotide repeat sequence in the non-coding region of the dystrophia myotonica-protein kinase (DMPK) gene. The abnormal expansion of CTG affects RNA transcription and splicing. This toxic RNA further impacts the function of chloride ion channels in skeletal muscles, leading to corresponding symptoms.
Toxic DMPK and reduced levels of the blind muscle-like RNA-binding protein (MBNL) have been identified as the genetic cause of DM1.
Research from the University of Valencia has found that miRNA is overexpressed in muscle biopsy tissues from DM1 patients and in various animal models with DM1. This reveals a significant link between miRNA and the mechanisms of DM1. In the pathogenic mechanism of DM1, miRNA affects the synthesis of MBNL protein by binding to mRNA, leading to a reduction in its levels, which in turn causes pathogenic mislocalization events in DM1 myoblasts.
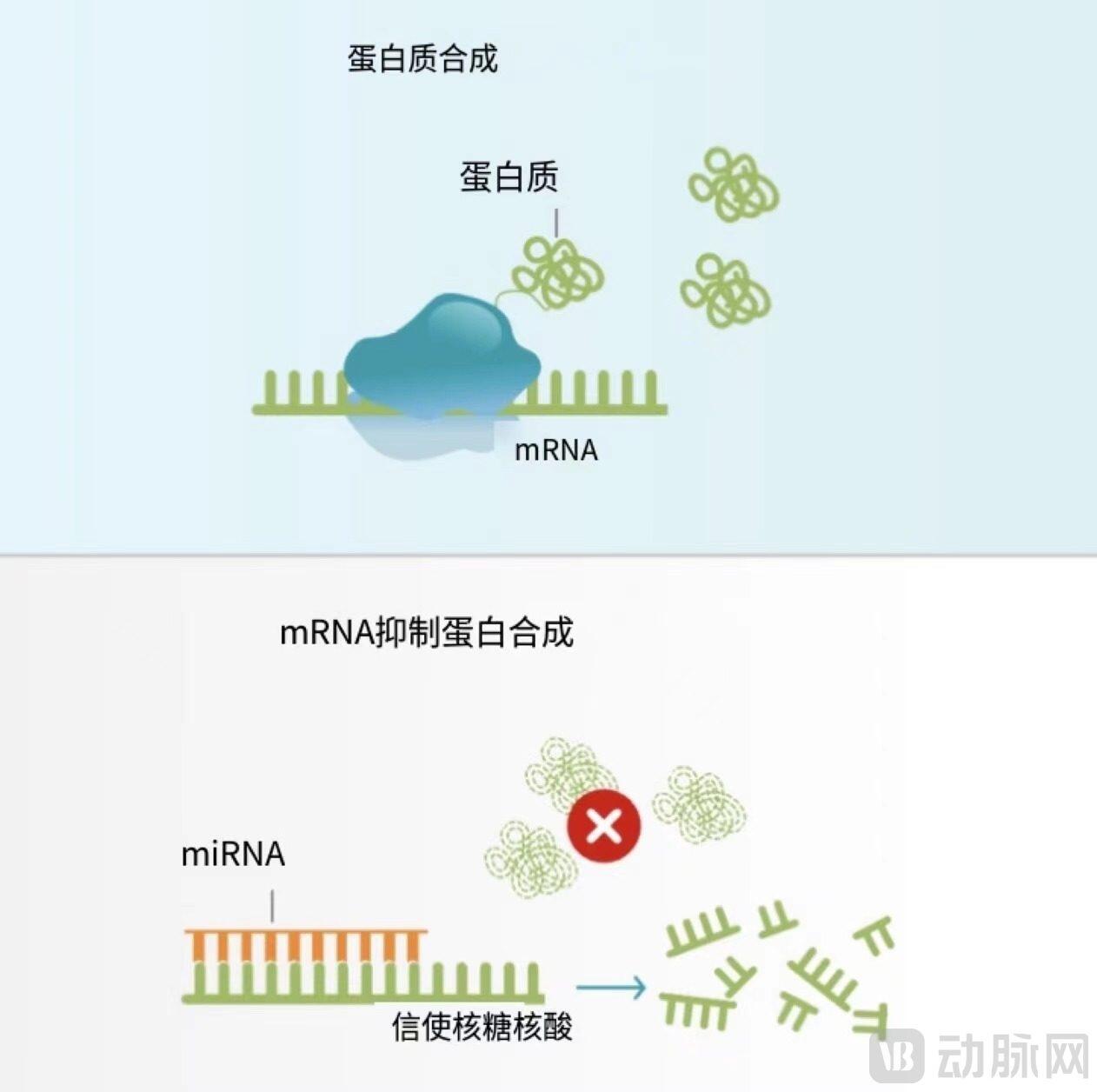
Normal Protein Synthesis (Top) / miRNA Inhibition of Protein Synthesis (Bottom)
Image source: ARTHEx Biotech
A common method to inhibit miRNA is the use of synthetic single-stranded oligonucleotides (known as antimiRs). The nucleotides composing antimiRs are chemically modified to enhance their binding affinity, stability, and biodistribution. These antimiR sequences are complementary to the target miRNA sequences, binding to the miRNA and sequestering, blocking, or degrading it to achieve the effect of enhancing MBNL protein levels.
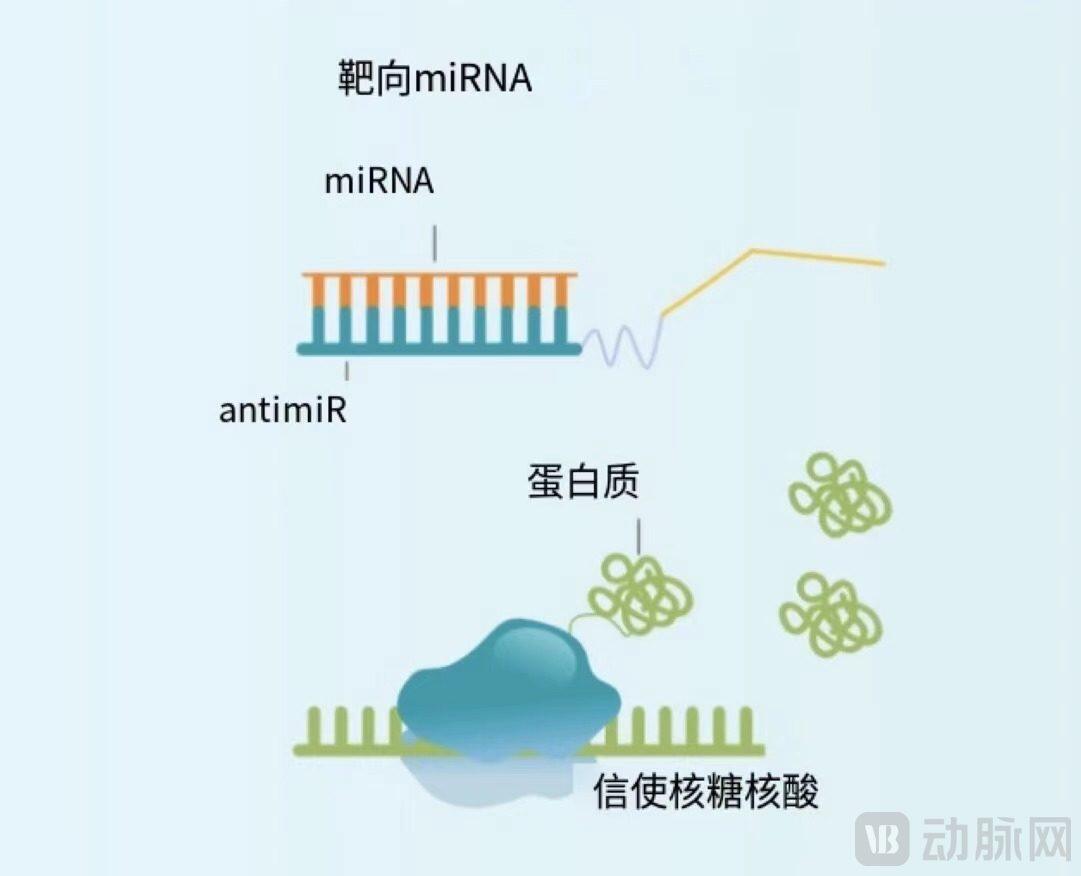
Principle of antimiR action using antimiR
Source: ARTHEx Biotech
In preclinical trials of HSALR mice, anti-miR was able to increase MBNL protein expression, reduce toxic DMPK lesions, and improve splicing defects.Reduce muscle stiffness and improve grip strength。
Based on the results of preclinical trials, the ARTHEx team has developed the anti-miR drug ATX-01, targeting miRNA 23b. This novel therapy hasA unique dual-action mechanism that can inhibit miRNA to increase MBNL levels while releasing MBNL from toxic DMPK foci.。
ATX-01 was granted Orphan Drug Designation (ODD) by the FDA in 2022.
Orphan drugs are products granted by the FDA for the diagnosis, treatment, or prevention of rare diseases that affect fewer than 200,000 people in the United States. Those granted orphan drug designation by the Office of Orphan Products Development (OOPD) are entitled to corresponding incentives:

Charting: VCBeat
In addition, ATX-01 has received confirmation and scientific advice from the European Medicines Agency (EMA) regarding its CTA data requirements. ARTHEx plans to submit its U.S. Investigational New Drug (IND) application and European Clinical Trial Application (CTA) in 2023.
ATX-01 will undergo lead compound selection in three different phases: the first phase is "in vitro" screening, the second phase is pre-selection, and the third phase is "in vivo" screening. Currently, both "in vitro" and "in vivo" tests show good potential. Meanwhile, no limiting toxicity was found in the toxicology studies of ATX-01 in minipigs and non-human primates.
In 2019, ARTHEx Biotech, supported by InVivo Capital, AdBio Partners, and CDTI-Innvierte, was established as a spin-off company from the University of Valencia.

Source: ARTHEx Biotech
Beatriz Llamusi, Co-founder and Chief Scientific Officer of ARTHEx Biotech, holds a Ph.D. in Biochemistry. She worked as a postdoctoral researcher at the Translational Genomics Laboratory of the University of Valencia and INCLIVA Health Research Institute. She has published over 20 related papers, focusing on the molecular basis of pathogenesis and potential therapeutic approaches, ranging from neural regeneration in the spinal cord to rare diseases, particularly myotonic dystrophy (DM). Based on her research into the molecular foundations of diseases, Dr. Llamusi also dedicates herself to identifying drugs that suppress phenotypes. She has discovered five anti-DM1 compounds or repurposed drugs.
The other co-founder, Dr. Ruben Artero, is a professor of genetics at the University of Valencia and the leader of the UV Translational Genomics Laboratory. Since 2005, he has been leading the Translational Genomics Laboratory. The lab focuses on the molecular basis of muscle wasting in muscular dystrophy and uses cellular, animal, and bioinformatics approaches to identify appropriate therapeutic targets and candidate drugs. During his Ph.D., Dr. Ruben Artero discovered the Muscleblind gene, the founding member of the MBNL protein family. He has authored more than 50 papers related to the mechanisms of muscular dystrophy, pioneering research on MBNL function and regulation. Through his work in this field, he has obtained over 10 patents and served as a consultant for various companies. He is currently a member of the ARTHEx Biotech Scientific Advisory Board.
During the transformation of scientific achievements by the two co-founders, ARTHEx first developed a proprietary technology platform called ENTRY. ATX-01 was discovered through this internal discovery engine, the ENTRY platform.
This platform aims to identify and optimize novel miRNA modulators (antimiR) oligonucleotide compounds and ensure their preferential delivery to target tissues for treating diseases where miRNA is involved in the pathogenesis.
The application of the discovery engine will allow miRNA modulator therapies to be applied to a broader range of disease areas, especially for diseases with unmet medical needs, including gene-driven conditions such as DM2.
As RNA therapy is gradually being applied to more diseases, the pace of drug development is accelerating, and the market remains a blue ocean. More rare genetic disorders that were once "untreatable" are now seeing hope, and we also anticipate a "surge of rapid growth" in drugs for rare diseases.