
In recent years, computer-aided drug design (CADD) and AI-driven drug design (AIDD), and its applications in drug discovery and design are becoming increasingly widespread. Against this backdrop, XtalPi has developed an intelligent and automated innovative drug discovery platform.ID4Inno, including two major computing systems: high-precision computational chemistry platformID4Gibbs And Artificial Intelligence Drug Discovery PlatformID4Idea, covering three major modules: intelligent computing, automated experiments, and expert experience. The platform explores a broader chemical space with higher efficiency and lower costs, significantly reducing the number of wet lab experiments and accelerating drug discovery.Design−Make−Test−Analyze(DMTA) Cycle.
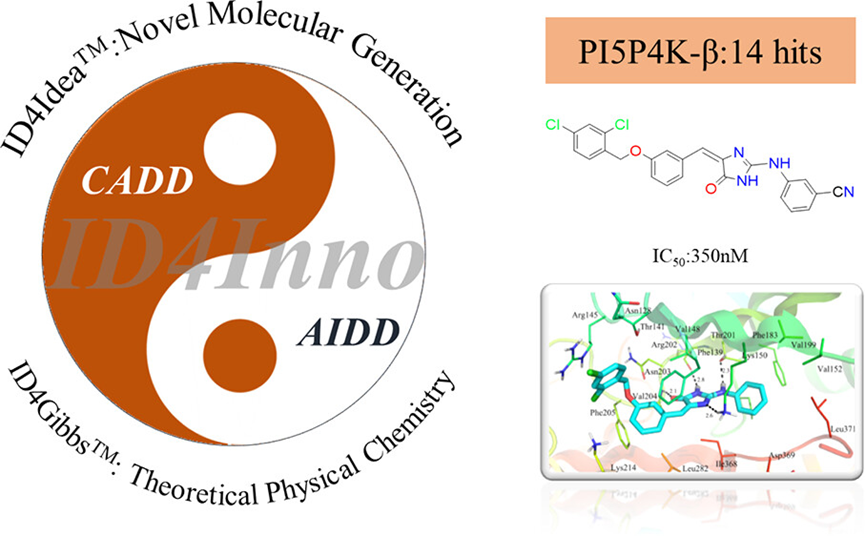
Recently, the team of XtalPi led by Lei Fang published in the core journal of computational chemistry from the American Chemical SocietyJournal of Chemical Information and ModelingPublished an article titled“Hit Identification Driven by Combining Artificial Intelligence and Computational Chemistry Methods: A PI5P4K-β Case Study”The research paper [1]. The team inID4InnoA workflow that combines artificial intelligence and computational chemistry to identify hit compounds has been developed on the drug discovery platform. This method features the innovativeness of artificial intelligence, the efficiency of computational chemistry, and the high performance of cloud computing, and has shown great potential in targeting.PI5P4K-βhas been well validated in the field of anti-cancer drugs.
Based onID4InnoThe platform's pioneering lead compound discovery process is shown in the figure.-1As shown. The first is virtual screening (VS): First, use molecular docking to quickly predict the binding mode of the target with small molecules and evaluate their affinity using a scoring function; then, based on semi-empirical quantum methods.PM7The calculated binding energy was screened to improveVSPositive rate; followed by two roundsMM/GBSACalculate and screen compounds with lower binding energy; finally, through manual inspection, select compounds with reasonable conformations and interaction patterns for absolute binding free energy calculation, and determine based on the calculation results.116A candidate compound. Considering the synthetic difficulty of subsequent structural modifications, the author purchased17A compound was tested and verified through biological experiments. The test results showed,9A compound in20 μMHigher than when50%The inhibition rate, with a screening success rate of53%. Among them, the compound with the highest activityvs1, half maximal inhibitory concentration is0.80 μM(Figure-2)。
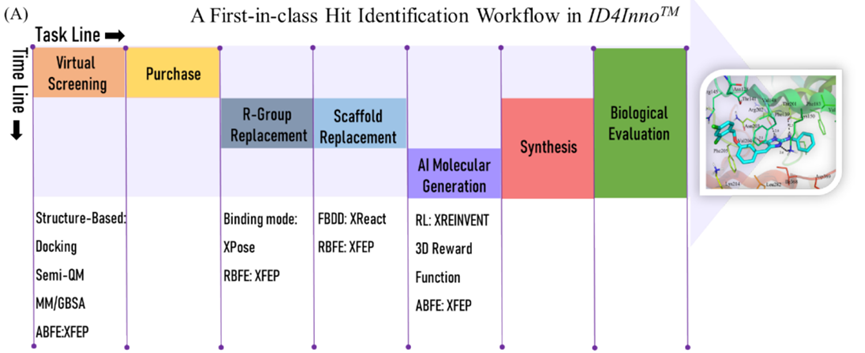
Figure-1: Based onID4InnoThe platform's pioneering lead compound discovery process.
Based onXPosePredictedvs1AndPI5P4K-βThe combination mode, the authorvs1ApplicationRA series of small molecules were designed using a group substitution strategy, andXFEPMethod to predict their binding ability with target proteins. Subsequently, some compounds were synthesized and tested for theirPI5P4K-βEnzyme inhibitory activity. Through structure-activity relationship analysis, it was found that the compounds obtained after structural optimization in the ring region showed activity comparable tovs1Similar,XFEPThe predicted relative binding free energy can effectively guide the optimization of substructures inside pockets far from the solvent region (Fig.-3)。
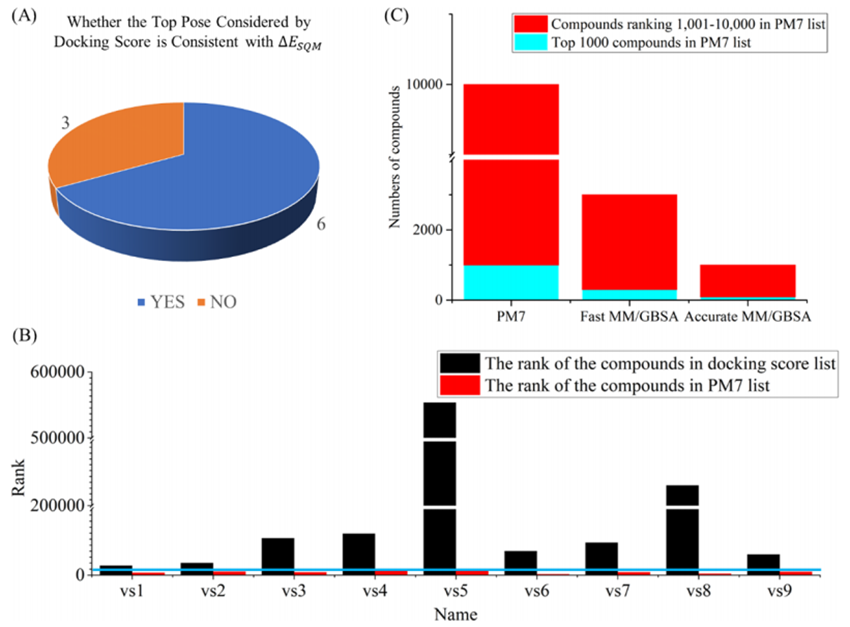
Figure-2:(A)ΔESQMComparison of the correlation with the highest scoring binding mode.B) Through docking scoring andΔESQMRank the highest binding modes of positive compounds.(C) PM7Before10000A compound inMMGBSASteps andPM7Comparison of Step Distribution.
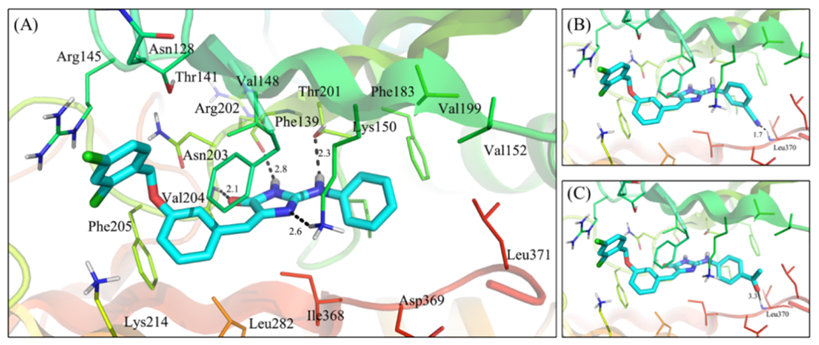
Figure-3: (A) byXPosePredictedvs1The binding mode.(B)d5The binding mode.(C)d8The binding mode.
ConsideringXPoseCombining Pattern Prediction andXFEPThe guiding significance of affinity prediction in the cyclic region is relatively weak, and the author adoptsID4IdeaInternal Methods of the ModuleXReactGenerate structures for this region. Select aniline imidazolone as the core scaffold (Fig.-4), substituent groups are limited to25A heavy atom and contains1-3A ring. In the generated molecules, the ligand efficiency is less than-0.3Classify molecules, between each pair of molecules in all categoriesRogers- TanimotoThe dissimilar distances are all no greater than0.15Select one compound from each class of compounds for stability verification based on molecular dynamics simulations. After manual inspection, absolute binding free energy filtering, and other steps, the final selection was made and synthesized.4A compound, wherein3A compound'sIC50The value is within the acceptable range. This method has successfully expanded the new framework structure space to12A compound.
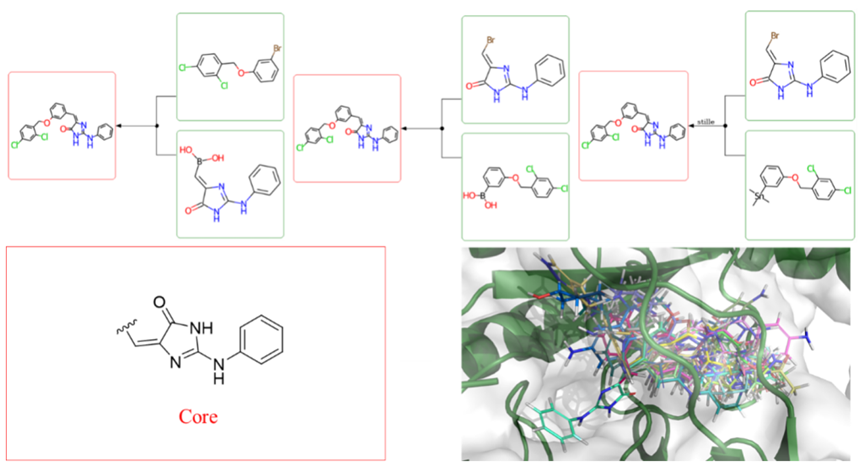
Figure-4: XReactProcess Example.
In order to explore more active structural space, the author appliedID4IdeaModuleXREINVENTPerform reinforcement learning to generate new molecules. The pre-trained generative model required for this method(Called the prior model)It is inChEMBLTrained on the dataset, it has a resemblance toREINVENTThe ability to generate and potentially sample compounds from a vast chemical space. Using a smaller set of compounds on this prior model for300Stepwise transfer learning, then based on multi-parameter objectives500Step reinforcement learning. The reinforcement learning process selects a three-dimensional pharmacophore constraint scoring function as the reward and punishment index. The author usesXREINVENTTwo rounds of molecular generation were designed. In the first round of reinforcement training, only the pharmacophore model and molecular weight were considered, and in the second round, compounds were added.vs1The substructure as a restriction to generate more similaritiesvs1Molecular structure. The final generation of5594The number of effective molecules in a molecular structure reaches99.41%, Novel Structure Accounts For86.5%. Using the aforementioned virtual screening process to filter all generated molecules, two novel scaffold structures were ultimately synthesized and obtained, which are effective againstPI5P4K-βThe inhibitory activity is less than20 μM。
In summary, the virtual screening,RStrategies such as group substitution and molecule generation, after biological experimental validation, ultimately achieve coverage encompassing14A New Framework26One TargetedPI5P4K-βThe budding compound (IC50<20 μM), wherein the compoundd5with the highest activity,IC50Value reaches350 nM。

Figure-5:14One TargetedPI5P4K-βChemical Structure of Active Compounds.
Summary:
Developed by XtalPiID4InnoAs an intelligent drug discovery platform that integrates smart algorithms, robotic experiments, and expert experience, it provides personalized one-stop computational services for new drug development.https://www.xtalpi.com/solution). The author of this paper is based onID4InnoThe platform has developed a workflow that combines artificial intelligence and computational chemistry, saving time and costs in discovering active lead compounds and demonstrating the potential application value of artificial intelligence methods in new drug development.
References:
【1】Wei L, Xu M, Liu Z, et al. Hit Identification Driven by Combining Artificial Intelligence and Computational Chemistry Methods: A PI5P4K-β Case Study. J Chem Inf Model. 2023, 63(16), 5341-5355.