Baidu and BioMap's HelixFold-Single: An MSA-Free, High-Efficiency Protein Structure Prediction Tool Based on Protein Language Models

BioMap
Developer of Innovative Drug R&D Platform


Compiled by Wang Cong
Editor | Wang Duoyu
Typesetting丨Shuicheng Wen
ProteinBasically involved in all biological processes, playing a crucial role in living organisms. The structure of proteins is highly correlated with their functions in biological processes. Determining protein structures to understand their functions can make significant contributions to the life sciences.
In recent years, based onArtificial Intelligence(AI)The protein structure prediction technology has made significant progress in prediction accuracy, showing great promise in the fields of drug and vaccine development. In particular, the technology developed by DeepMind, a subsidiary of Google,AlphaFold2Pushing protein structure prediction to a new peak.
Mainstream protein structure prediction tools heavily rely onMultiple Sequence Alignment(MSA)The co-evolutionary information extracted from it. MSA can be simply regarded as protein chains similar to the target protein chain sequence, and the co-evolutionary information of MSA is related to the protein sequence, which is crucial for predicting its structure. However, over-reliance on MSA has become a bottleneck for various protein-related tasks because searching for MSA is very time-consuming — it takes several tens of minutes for one protein, which is detrimental for tasks such as protein design.
Therefore, designing an accurate and efficient protein structure prediction method that does not rely on MSA has the potential to promote discoveries in protein research, thereby accelerating the development of new drugs for treating various diseases.
October 9, 2023BioMapSong LeDr. UnitedBaidu Natural Language Processing DepartmentResearchers at NatureSub-publicationNature Machine Intelligence Published an article titled:A method for multiple-sequence-alignment-free protein structure prediction using a protein language model Research Paper.
The study developed a method namedHelixFold-SingleEnd-to-endProtein Structure PredictionMethod, a method that combines large-scaleProtein Language Model(PLM)AndAlphaFold2Superior geometric learning ability, not dependentMultiple Sequence Alignment(MSA), only from the primary structure(Amino Acid Sequence)Predicting the three-dimensional coordinates of atoms to achieve accurate prediction of protein structures. Moreover, HelixFold-Single outperforms the current mainstream MSA-based protein structure prediction tools.(AlphaFold2AndRoseTTAFold)Takes much less time in therapeutic protein(Peptides, Antibodies, and Nanobodies)Has great application potential in design.

The research team believes that, on a large scale,Protein Language Model(PLM)Can be used asMultiple Sequence Alignment(MSA)An alternative solution to learn co-evolutionary knowledge for predictions that do not rely on MSA.
The MSA-based method uses information retrieval technology to explicitly capture the co-evolution information of target proteins from protein sequence databases, while the PLM-based method embeds co-evolution information into large-scale model parameters during training and performs implicit retrieval through model inference, where PLM can be regarded as a protein knowledge base. Compared with MSA, the PLM-based method is more efficient in information retrieval.
In the past few years, large language models(LLM)Achieved great success in the field of natural language processing, such as the currently popularChatGPT. And now, there have been some studies attempting toProtein Language Model(PLM)Applied toProtein Structure Prediction, but the predictive effect is still not satisfactory.
AffectedProtein Language Model(PLM)AndAlphaFold2Inspired by the progress, the research team proposed an end-to-end protein structure prediction tool that does not rely on MSA —HelixFold-Single。
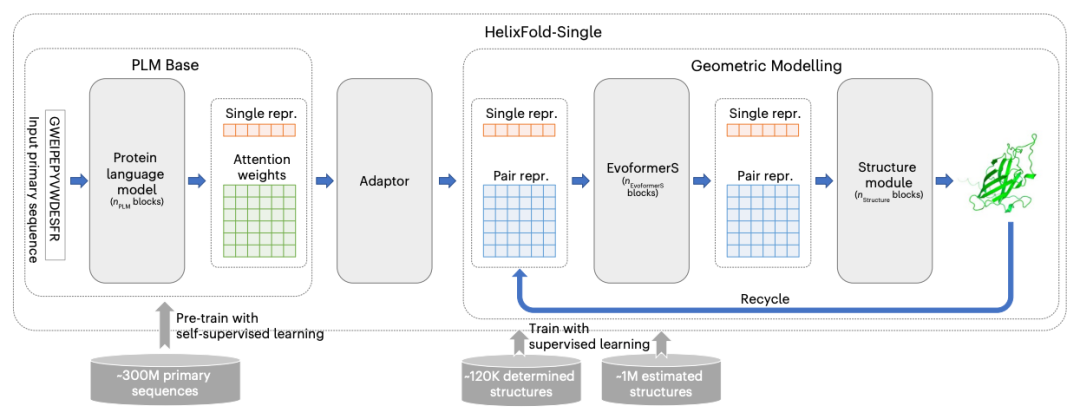
HelixFold-SingleThe model used in consists of two main components - a large-scale foundationProtein Language Model(PLM)And the basic components from AlphaFold2 for protein folding.PLM can encode the main structure into single representation and paired representation to learn domain knowledge.。Then, integrate the Evoformer and Structure modules in AlphaFold2 to process representations, learn geometric knowledge, and predict atomic coordinates. These two components are connected to provide an end-to-endDifferentiable Model(Differentiable Model)。
HelixFold-Single includes two training phases. In the first phase, a large-scale PLM is trained using millions of unlabeled single sequences through a masked language prediction task. In the second phase, the entire model is trained using protein structures composed of experimentally resolved and AlphaFold2-generated enhanced structures.
Next, the research team willHelixFold-SingleCompared with two mainstream protein structure prediction toolsAlphaFold2(Developed by DeepMind)AndRoseTTAFold(Developed by David Baker's lab at the University of Washington)Comparison on the CASP14 and CAMEO datasets.
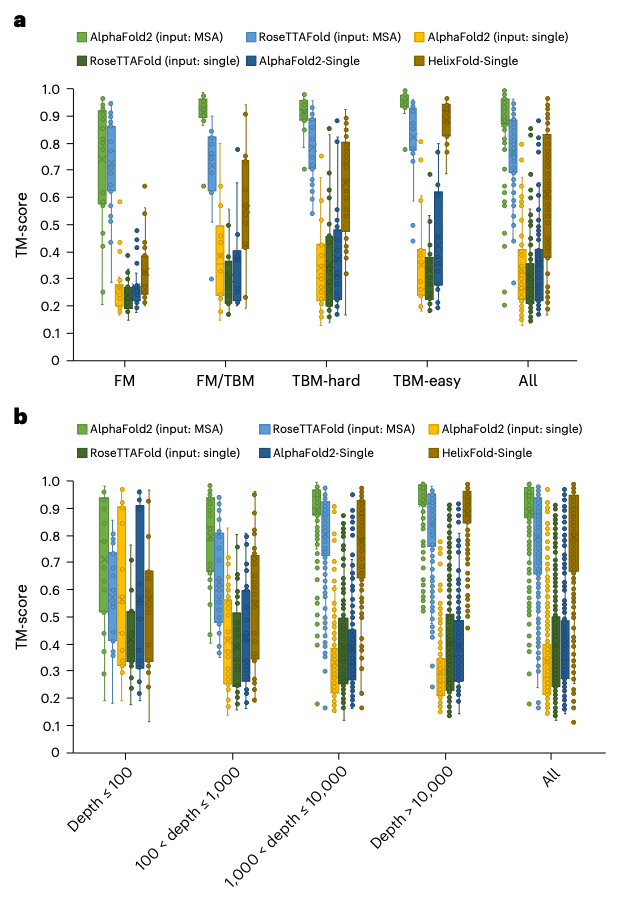
HelixFold-SingleAchieved with a sufficient number of homologous sequences on the proteinAlphaFold2AndRoseTTAFoldConsiderable accuracy. The research team also analyzed the performance of HelixFold-Single on targets with different numbers of homologous sequences: HelixFold-Single is able to provide accurate structure predictions for most targets, especially those with large homologous families.
In addition, mainstream protein structure prediction tools based on MSAAlphaFold2AndRoseTTAFoldMethod comparison,HelixFold-SingleIt has a great advantage in prediction efficiency, takes much less time, and can be applied to protein-related tasks that require a large number of predictions.
Specifically, the research team explored HelixFold-Single in various types of representative proteins(Including peptides, antibodies, and nanobodies)The predictive accuracy on the platform was assessed to evaluate its potential application in therapeutic protein design. The results indicate that HelixFold-Single performs well in predicting the flexible regions of these proteins, highlighting its advantages in such applications.