Genentech Refines the Cancer-Immunity Cycle Framework: Implications for Immunotype-Guided Therapeutics

Genentech
Pharmaceutical R&D Manufacturer
Tumor-The immune cycle provides a framework for understanding the series of events that generate an anti-cancer immune response. It emphasizes the iterative nature of the response, whichTThe killing of tumor cells by cells initiates subsequent antigen presentation andTCell stimulation, maintenance of active immunity, and adaptation to tumor evolution. Any step in the cycle may become rate-limiting, preventing the immune system from controlling tumor growth.
Recently, scientists from Genentech published a review titled "The cancer-immunity cycle: Indication, genotype, and immunotype" in the journal Immunity.Building on a decade of basic research, the cancer-immunity cycle continues to be updated. Just as perspectives on dendritic cells sustaining anti-tumor immunity have evolved, so too has the understanding of checkpoint inhibition mechanisms undergone iterative progress. It is also noted that the tumor microenvironment not only suppresses anti-cancer responses but may also enhance anti-tumor immunity. Additionally, the review discusses the importance of examining "immune types" in tumor immune phenotypes. While these new insights add complexity to the original framework of the cancer-immunity cycle, they also provide new targets for research and therapeutic intervention.
Preface
Despite decades of research on the role of the immune system in cancer, the current surge of interest is primarily driven by clinical observations. These observations stem from the outcomes seen in patients treated with immune "checkpoint" CTLA4 and PD-L1-PD-1 antibodies, even in cases of T-cell exhaustion under PD-L1-PD-1 axis conditions. In 2013, researchers introduced the concept of the cancer-immunity cycle (CI cycle), which indicates that T cells neither respond nor function independently but exist within a series of steps, some of which are even exogenous to both the immune system and cancer (see figure below).
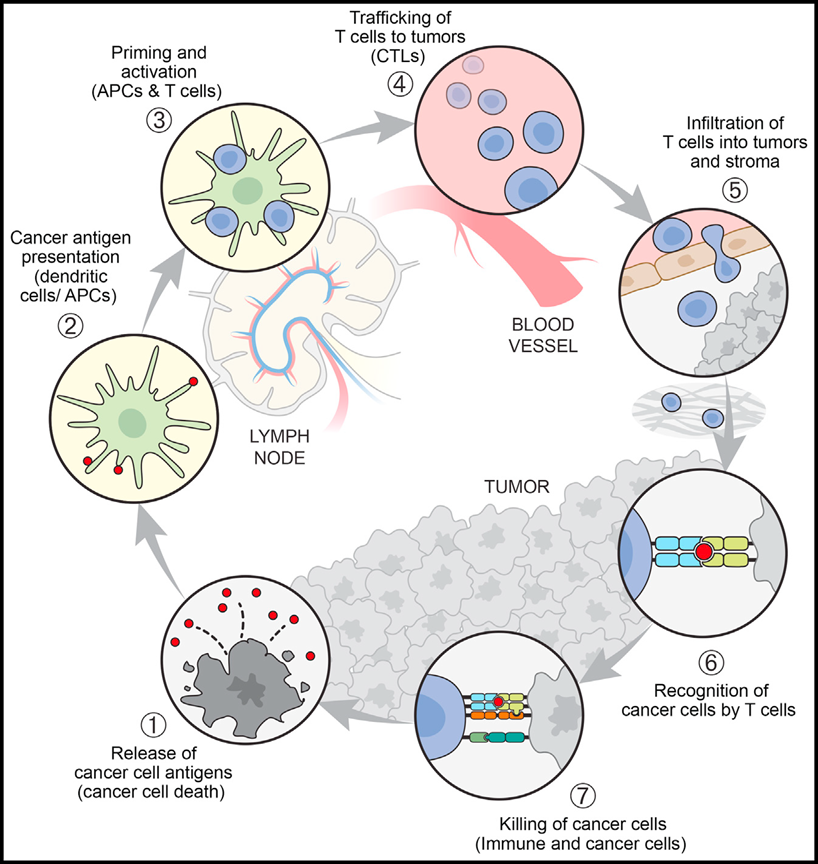
These steps are interrelated in a cycle, meaning that (1) any single step may potentially become the rate-limiting factor for generating optimal immunity, and (2) successful anti-cancer immunity has the potential to self-amplify during the response process. Even therapeutic strategies that create "synthetic immunity," such as adoptive cell therapy, antibody-linked immune cells, or CAR-T cell therapy, must function within the context of the cancer-immunity cycle.
CI Cycle Framework and Tumor Environment
The basic framework of the CI cycle has remained unchanged since its introduction, including subsequent modifications to emphasize that blood-derived T cells must often cross stromal barriers before reaching the tumor itself.However, according to recent research, some important new concepts need to be emphasized.
Even within individual cancer indications, tumors can still be considered to have different immune phenotypes or "immune types." The three classic immune types (immune-inflamed, immune-excluded, and immune-desert) are defined as tumors with abundant immune infiltration, tumors where T-cell infiltration is limited to the stroma rather than the tumor parenchyma, and tumors that do not exhibit immune infiltration (see figure below).
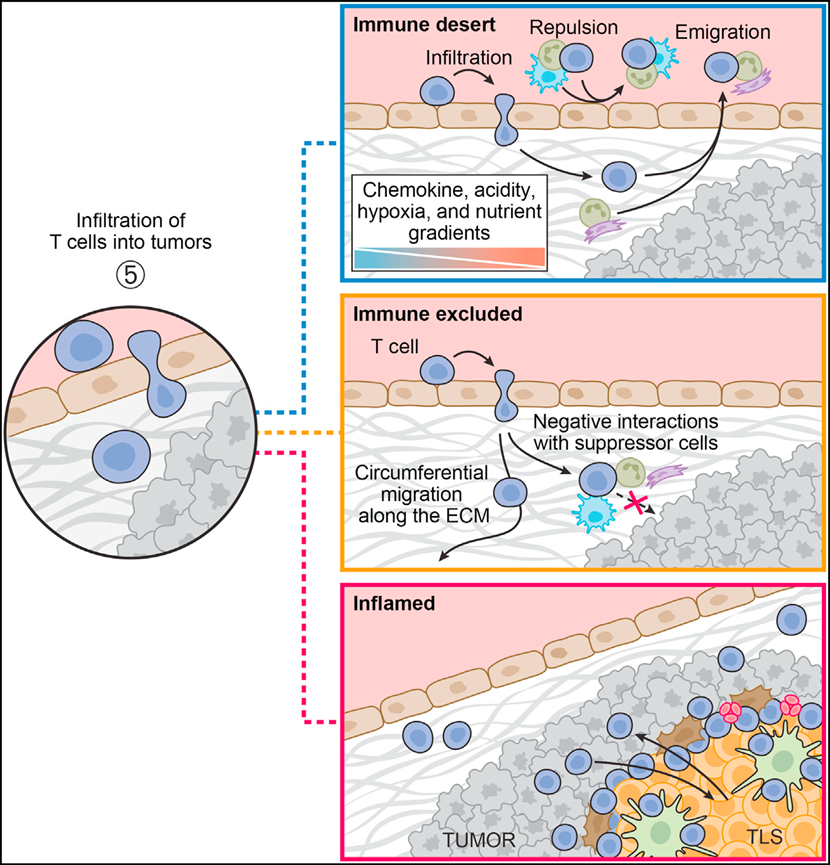
Although immune types may oversimplify the dynamic characteristics of tumors and can also change during tumor evolution or through therapeutic interventions, they do indeed represent a useful, mechanism-based classification system. Immune types appear with varying frequencies across different indications. For example, untreated prostate cancer, colorectal cancer, and melanoma most frequently exhibit desert, excluded, and inflamed types, respectively. However, importantly, all three immune types also occur in different patients within these indications. Therefore, immune types remain a useful framework for understanding the mechanisms of response and non-response and for guiding future research. Identifying the factors that lead to the formation of excluded or desert immune types will aid targeted discovery efforts and hopefully significantly increase the percentage of responding patients. Since the mechanisms responsible for these immune types are crucial for developing better immunotherapies,Referring to tumors simply as "hot" (presence of T cells) or "cold" (lack of T cells) is an oversimplification and, in fact, misleading.For example, immune-excluded tumors have T cells, but T cells are spatially restricted from tumor cells, so they are usually resistant to checkpoint blockade.
In view of the current significant progress, it is necessary to revise the initial perspective of the CI cycle to incorporate the TME, particularly the crucial role of DCs in regulating and sustaining anti-tumor T-cell responses. As shown in Figure 3, this is best illustrated by the "sub-cycle" that occurs at the tumor site when dLN-derived T cells enter the tumor (in step 5 of the CI cycle).
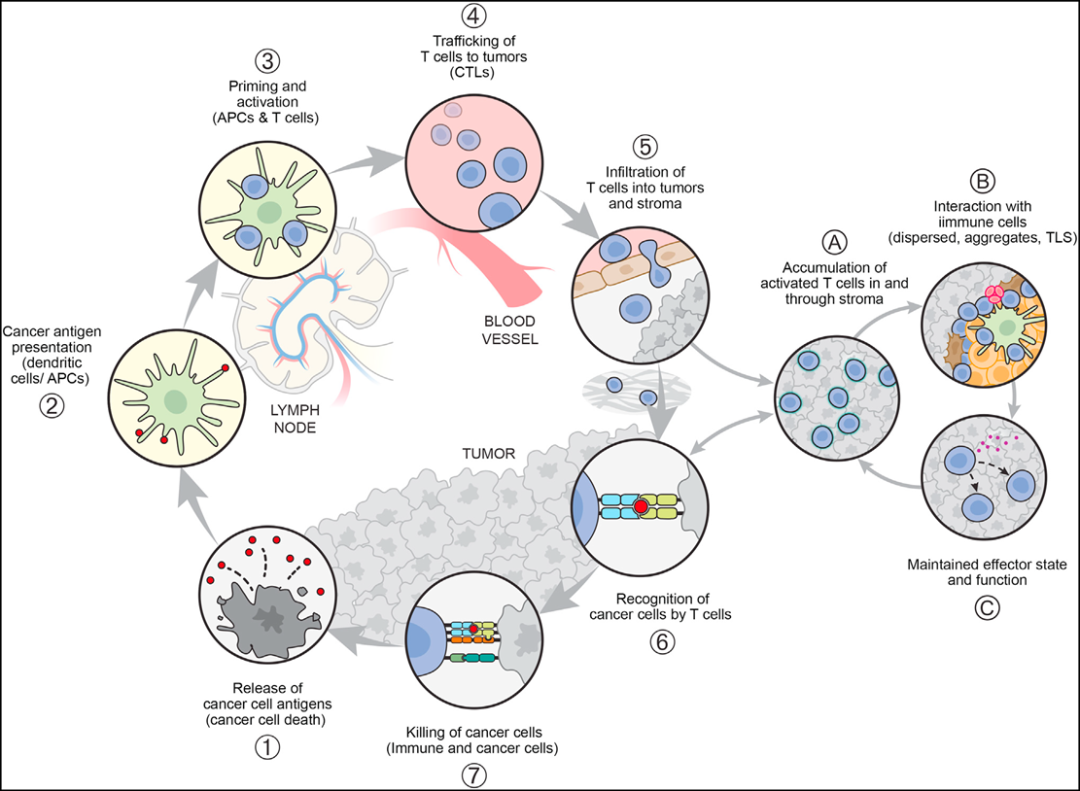
These T cells will encounter antigen-presenting cells (especially DCs) dispersed within the tumor parenchyma, tumor-associated lymphoid aggregates, or morphologically identifiable TLS. The T cells can then expand and differentiate (e.g., into effector, memory, or exhausted states), leading to the direct killing of tumor cells and potentially initiating a local TME "vortex" of the CI cycle. This perspective underscores the crucial and complex role of the TME in both supporting and suppressing cancer immunity (CI cycle steps 5, 6, and 7). It is conceivable that this role implies a series of new potential therapeutic targets. The figure below highlights some known molecules or interactions influencing T cell behavior throughout the CI cycle and sub-cycles, while exemplifying the range of potential intervention sites.
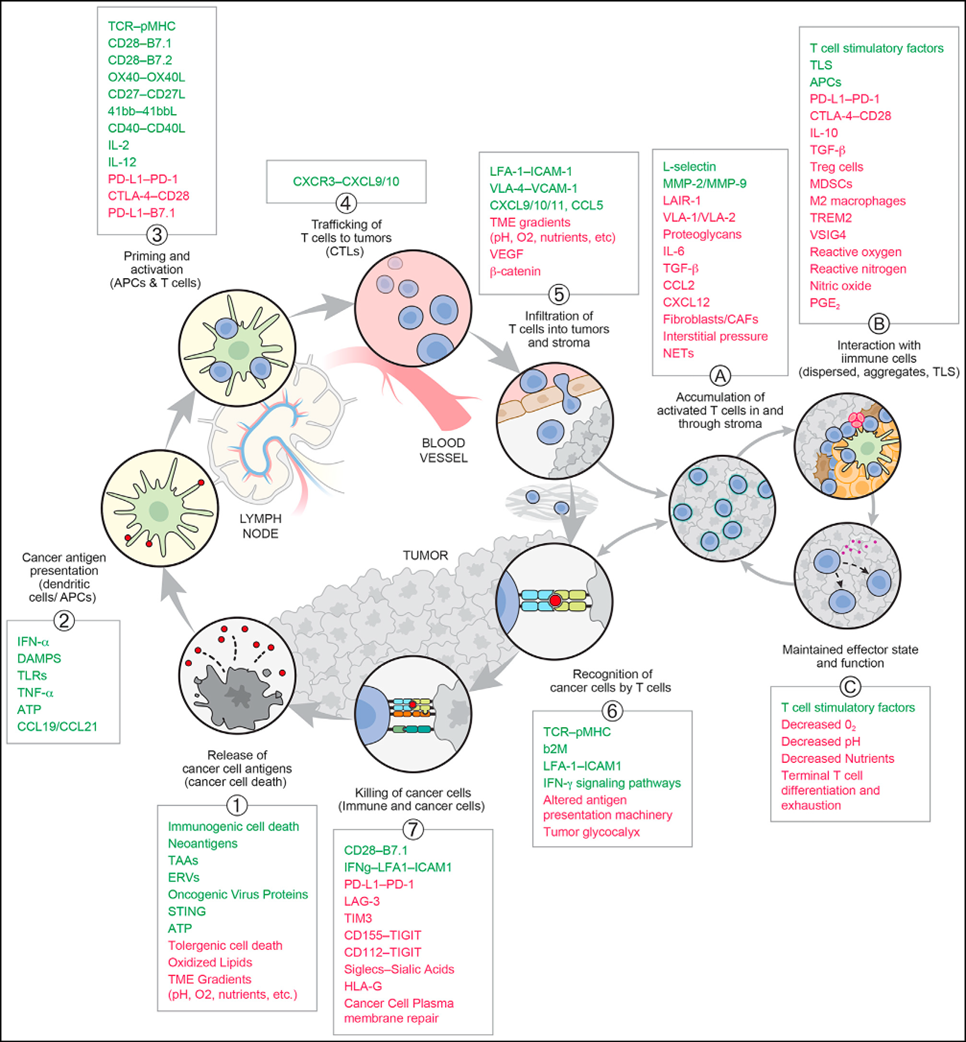
Immunosuppression of Cancer-Associated Fibroblasts
The conceptual advance in immunosuppression may lie in the recognition that the fibroblast compartment, cancer-associated fibroblasts or "CAFs," could play a pivotal role. CAFs develop from fibroblasts after exposure to activating signals from tumor cells as well as alterations in oxygen and metabolite gradients and availability, and they play a crucial role in establishing the stromal architecture of the TME. CAFs exhibit significant functional pleiotropy, influencing various hallmarks of cancer such as tumor initiation, metabolism, progression, and metastasis, anti-tumor immunity, angiogenesis, drug penetration, and therapeutic response. These functions are partly due to their decisive role in forming the complex matrix environment and the tissue mechanisms of tumor growth and metastasis.
Despite decades of research on CAFs, the field still lacks a consistent framework to capture cellular subpopulations and states, cell surface markers, lineage-defining transcription factors, developmental origins, localization patterns, and functions. The emergence of single-cell omics technologies, particularly single-cell RNA sequencing (scRNA-seq), has rapidly advanced the understanding of CAFs, providing new markers, subgroup identities, and granularity for tool generation in mechanistic studies. Three main classes of CAFs have been observed in most human solid tumors: myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs). myCAFs represent a prominent CAF subtype in most human solid cancers, especially in advanced tumors, producing large amounts of extracellular matrix (ECM) and other fibrosis-related molecules. The CAF pattern of stromal architecture influences cancer cell invasiveness, immune cell infiltration, angiogenesis, organ stiffness, and drug penetration. Additionally,myCAF is also immunomodulatory, showing the potential to suppress and regulate CD8 T cells and other immune cells, capable of producing various cytokines and chemokines, while iCAF secretes IL-6 and chemokine ligands CCL2 and CXCL12, exerting an immunosuppressive effect.These cells dominate the CAF compartment in selected metastatic environments. Antigen-presenting CAFs are similar to iCAFs in expressing immunomodulatory factors, also express relatively high levels of MHC class II molecules, and induce the recruitment of regulatory T (Treg) cells.
Immunosuppression of Bone Marrow Cell Compartments
Myeloid cells are the most abundant cell type in solid cancers other than cancer cells themselves, with monocytes and immature myeloid cells (also known as myeloid-derived suppressor cells or MDSCs) accounting for nearly half of all cells in the tumor microenvironment. Neutrophils and dendritic cells (DCs) are also present in most human solid cancers but comprise a much smaller proportion (<10%) of the tumor myeloid compartment. Myeloid cells thrive in the TME partly due to the large amounts of growth factors, nutrients, cytokines, and chemokines secreted by tumor cells (e.g., M-CSF/CSF-1, IL-6, GM-CSF, G-CSF, CCL2, CCL5).
TAMs in tumors exert both pro-tumor and anti-tumor functions and contribute to multiple hallmarks of cancer. Moreover, TAMs exhibiting pro-tumor characteristics appear to significantly outnumber those with anti-tumor functions, but the exact phenotypic traits and functional contributions of TAM subtypes remain incompletely understood. Generally, the anti-tumor functions of TAMs include killing and phagocytosing tumor cells, MHC class II antigen presentation, and expression of pro-inflammatory cytokines. The pro-tumor functions of TAMs encompass the expression of factors promoting angiogenesis, ECM remodeling, cytokine expression, and suppression of anti-tumor immunity through the induction of Treg cells. TAMs express PD-L1 and other molecules that inhibit T-cell responses to tumors. TAMs also secrete factors that promote blood vessel growth and tumor cell metastasis.
Given the absolute size, developmental complexity, and functional impact of the macrophage compartment on cancer cells, the tumor microenvironment (TME), and anti-tumor immunity, it is reasonable to suggest that breaking the efficacy ceiling in cancer therapy may require strategies targeting myeloid cells.Currently, many methods have been evaluated in clinical trials, such as the depletion of total macrophages, but so far without success. However, other novel therapies are under development, including selectively depleting pro-tumor TAMs, directly inhibiting their pro-tumor functions, or reprogramming TAM subtypes from a pro-tumor state to an anti-tumor state. Additionally, potent innate activators, such as Type I IFN, are being utilized through targeted delivery or by inducing in situ formation within tumors. Since the activation of innate immunity is key to initiating the CI cycle both systemically and intratumorally (in cases of sub-cycles), this strategy should attract significant interest.
Tumor Immunosuppression
In addition to the numerous suppressive mechanisms of the TME, tumor cells themselves also possess the ability to limit T-cell immunity. While several mechanisms have been described in preclinical models, few have been clinically validated or offer new therapeutic targets. For instance, in melanoma models, the activation of β-catenin signaling is associated with an immune desert and resistance to checkpoint inhibitors. Although it remains unclear whether this is due to a tumor-specific defect or failure of NK cell infiltration, the effect has been attributed to insufficient secretion of T-cell chemokines. Prostaglandin E2, a regulator of T cells and other immune cells, is linked to the activation of the cyclooxygenase pathway in tumors and resistance to immunotherapy. Similarly, tumors carrying IDH1 or IDH2 mutations (particularly gliomas) overproduce 2-hydroxyglutarate, thereby suppressing T-cell function.
Activation of carcinogenic pathways may also directly or indirectly counteract T-cell immunity. For example, increased Ras/MAPK signaling reduces the expression of MHC class I gene products, thereby decreasing the susceptibility of tumor cells to T-cell attacks. In rare cases, tumors are protected by limiting the cytotoxicity of T-cell effector release of IFN. Finally, tumor cells can protect themselves from T-cell killing by rapidly repairing plasma membrane pores generated when perforin is released from T-cell granules.
TME: Immunostimulation of DCs
DCs Remain Indispensable in the CI Cycle Due to Their Unparalleled Ability to Initiate and Amplify Antigen-Specific CD4 and CD8 T Cell Responses. Over the past decade, significant progress has been made in defining and functionally characterizing various DC subsets and populations. The conventional DC1 (cDC1) subset remains the most crucial initiator of CD8 T cell tumor immunity, at least partly reflecting their ability to migrate from the tumor bed to the dLN, cross-present internalized tumor antigens on MHC class I molecules, and stimulate naïve CD8 T cells. These or other migratory cells may also "transfer" tumor antigens to dLN-resident DCs in some way, representing a second option for antigen cross-presentation to T cells on both MHC class I and II molecules. Additionally, two other general types of DCs exist, though their roles are less well-defined. cDC2 is typically associated with MHC class II molecule presentation and stimulation of CD4 responses. cDC3, also known as CCR7 DCs or mRegDCs, have been identified within tumors and in dLNs, are migratory, and can mediate immunostimulatory or regulatory functions depending on the microenvironment.
In the context of immunotherapy, PD-L1 expression on DCs plays a disproportionately important role in controlling T cell responses and is also a more effective predictor of response in human cancer patients than total PD-L1 expression (including tumor cell expression). Finally, possibly due to antigen presentation by DCs, T cell exhaustion within tumors may be controlled when the tumor itself is under control. Together, these concepts strongly suggest that,The role of DC in the TME is not limited to transferring antigens from the tumor to the dLN but also ensures the activation and expansion of antigen-specific T cells within the tumor itself.
TLSs in Tumors
Tertiary lymphoid structures (TLSs) are essentially primary LNs containing germinal center-like structures and have long been known to occur in tumors. However, their potential role in tumor immunity has only become clear in recent years. Human clinical studies have shown that responses to checkpoint therapy are often associated with the presence of TLSs in the tumor microenvironment (TME). Particularly, given the accumulating evidence that DCs within the TME may function in situ, clinical data suggest a functional link. By providing an organized LN-like structure for T cell stimulation, TLSs can serve as sites for the activation and expansion of T cells by tumor-associated DCs.
The enhanced relevance of TLSs to the TME introduces the concept that, in addition to immunosuppression, it can also exhibit immunostimulatory properties, and DC stimulation of T cells is not limited to secondary lymphoid organs (e.g., dLN) but occurs within the tumor itself. This activity may not be restricted to TLSs but distributed across the entire TME and intratumoral DCs (and possibly other antigen-presenting cells). The model suggests that, in addition to their recognized role after lymphatic migration to dLN, DCs can also stimulate T cells in situ.
CI Cycle Guides T Cell Differentiation and Function
T cells can be primed and further stimulated in both the dLN and the TME, raising important questions about T cell differentiation and trajectory control. The initial assumption that T cell activation and expansion occur exclusively in the dLN (Step 3 of the CI cycle) suggests that all subsequent characteristics of T cell function are determined at this site. Thus, whether T cells are destined to enter exhaustion, effector, or memory pathways would be determined by the conditions of antigen presentation in the dLN. However, as this simple assumption no longer appears to be correct, it is possible that only initiation or activation begins in the dLN, while terminal differentiation occurs at the tumor site (a "sub-cycle" of Step 5). It is also possible that all these activities occur simultaneously at both sites, and in some cases, TLS may serve as the site for T cell priming within the TME.
Recent studies have shown that geographically distinct DC populations can play different roles in the development and exhaustion of T cells. Although the identity and precise trajectory of the T cell populations involved remain unclear, an attractive model may be that tumor-specific T cells maintain dLN in a relatively pluripotent state, undergo terminal differentiation within the tumor, including the formation of tissue-resident memory T cells and central memory T cells, which may then recirculate. Regardless of the model, T cells are guided by intratumoral DCs to differentiate along effector, memory, or exhaustion pathways.
The Determining Role of Tumor Immune Typing
Currently, less than half of the patients achieve durable outcomes through immunotherapy, even with combination immunotherapy. Identifying predictive response characteristics or understanding mechanisms of resistance remain primary focuses in preclinical and clinical research.These factors may be intrinsic to the tumor or the TME, or they may reflect the patient's genetics, microbiome, metabolic, or pharmacological state, but in each case, they must reflect the location of the rate-limiting step in the CI cycle.PD-L1 Expression on Tumor Cells or Immune Cells (Particularly DCs) Remains the Most Useful Parameter for Patient Selection, but It Is an Imperfect Predictor, Possibly Because It Does Not Necessarily Indicate the Specific Rate-Limiting Step in the CI Cycle. Mechanistically, PD-L1 Expression Is Believed to Suggest That a Patient Has an Ongoing Antitumor Response, with IFN-γ Released by Effector T Cells in the Tumor Bed Leading to Surrounding Cells, Especially DCs, Which May Be Involved in Guiding the Terminal Differentiation of Newly Arrived or Locally Produced T Cells. Even if This Concept Is Correct and PD-L1-Positive Patients Do Have Preexisting Immune Responses, It Does Not Necessarily Mean That Blocking Coinhibitory Receptors Such as PD-1 Will Overcome the Rate-Limiting Step of the CI Cycle in a Specific Patient.
Host-Related Factors Influence Tumor Immunity
Host and environmental factors may influence the CI cycle and response to immunotherapy. A high polygenic risk score for vitiligo or psoriasis derived from germline SNPs is associated with longer OS under anti-PD-L1 monotherapy compared to chemotherapy. This suggests that the host's response to tumorigenesis correlates with predictive outcomes. Additionally, epigenetic factors such as chromatin structure regulate the expression of key immune-related proteins. Lastly, the impact of the gut microbiome on the immune system is well-established, but understanding its role (positive or negative) in the CI cycle will significantly enhance our understanding of the underlying mechanisms.
Concomitant medications also play a role in determining the outcomes of immunotherapy. In addition to the anticipated effects of lymphatic-absorbed chemotherapy, prior treatment with antibiotics that deplete the gut microbiota often has a negative impact. The effect of antibiotics may demonstrate the positive influence of the microbiota on anti-cancer immune responses. On the other hand, various tumor-targeted therapies, such as Ras-MAPK inhibitors and Cdk4/6 inhibitors, can enhance anti-cancer immune responses by increasing tumor antigen presentation or promoting T-cell function.
The Clinical Significance of CI Circulation and Its Repair
Immune checkpoint inhibition (ICI), particularly PD-L1/PD-1 therapy, has successfully transitioned from advanced stages to the perioperative setting, reducing post-surgical recurrence rates and altering the prognosis for specific tumor types. Randomized trials in the perioperative phase are currently underway. In melanoma, a neoadjuvant approach appears preferable to adjuvant therapy. Although the mechanistic basis of this effect has not yet been studied, applying the logic of CI cycling may predict that the neoantigen load during treatment allows checkpoint blockade to enhance T-cell responses, enabling subsequent surgery to reduce the overall tumor burden. This permits T cells, which were insufficient in generating a durable response during the neoadjuvant phase, to control tumor growth during adjuvant therapy. This suggests that tumor burden can be considered as both a rate-limiting factor before surgery and a rate-limiting factor for T-cell activity after surgery.
Currently, the optimal duration of ICI therapy and the issue of immune memory after treatment cessation have not been fully resolved. Although dynamic changes in the TME occur during ICI therapy, their correlation remains uncertain and requires further investigation both preclinically and in patients. Following recent advances in ICI therapy, re-challenge with PD-(L)1 treatment appears to be unrelated to clinical benefit, suggesting that the loss of response reflects the development of another rate-limiting step within the CI cycle.Although the mechanism of action of CLTA-4 remains uncertain and is associated with higher toxicity that many patients cannot tolerate, the only established ICI combination to date is PD-1 and CLTA-4 inhibition. Anti-CTLA-4 can play a role in promoting the initiation of new T-cell responses or depleting Tregs, with the potential to suppress anti-cancer T-cells. Thus, anti-CTLA-4 can act at two sites in the CI cycle (see figure below).
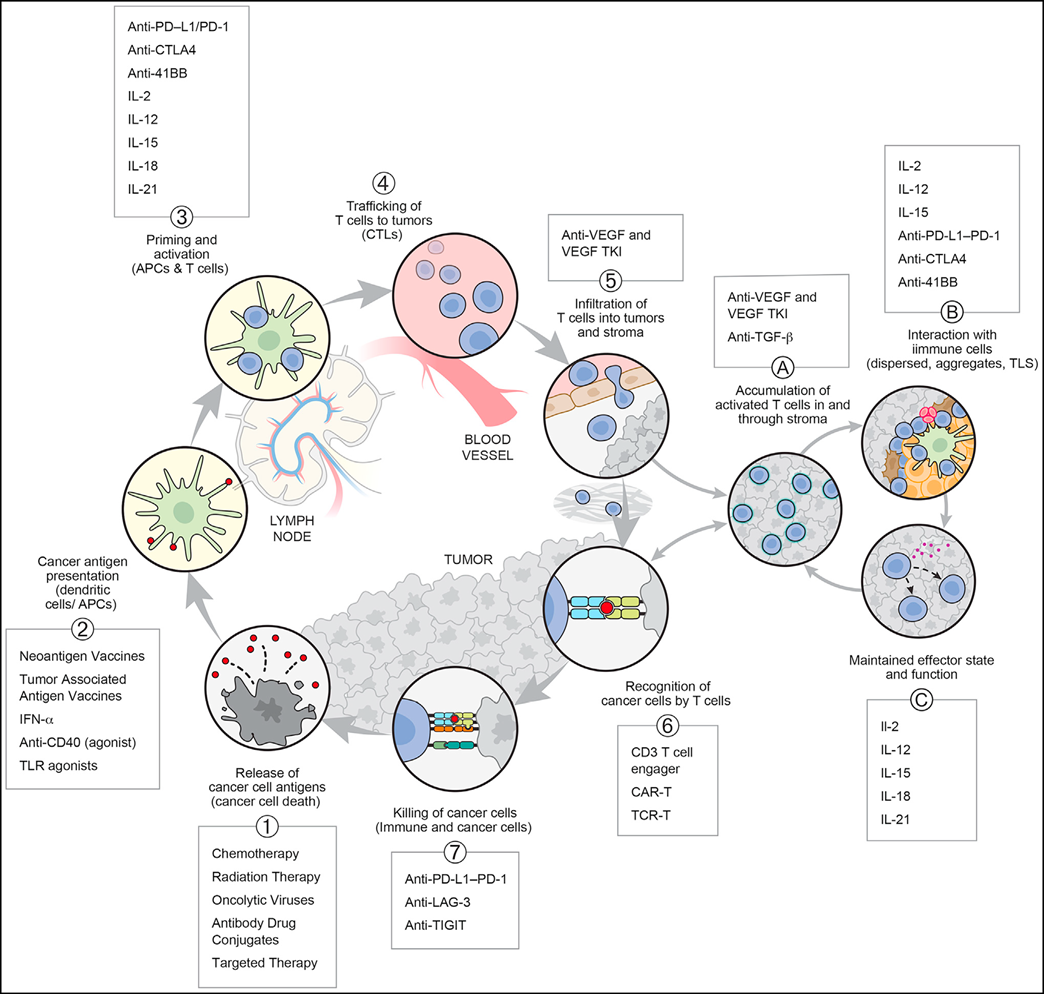
Another challenge limiting clinical progress is that many combinations are tested under suboptimal conditions, with small single-arm trials conducted in heterogeneous patient populations previously treated with immunotherapy. Many combinations that might be effective in specific clinical settings may have been prematurely discarded. Robust initial testing is highly desirable; if drugs without single-agent activity are to be developed, there must be a testable therapeutic hypothesis that can be evaluated in trials, so that important mechanistic and pharmacodynamic information can be obtained regardless of the trial's efficacy. This again brings us back to the development of concepts like the CI cycle: having a clear framework within which one can see the steps necessary to generate and sustain an effective anticancer response is critical for interpreting complex clinical outcomes.
Summary
Ten years after the publication of the CI cycle, its basic characteristics still accurately reflect the understanding of cancer immune responses. However, understanding each step of the cycle and their interconnections does not necessarily ensure comprehension of their mechanisms of action. It can be observed that due to detailed research, the initial mechanistic hypotheses—and even successful treatments, such as checkpoint inhibitor-induced reversal of exhaustion—have evolved. Additionally, it has been noted that T-cell activation is influenced not only in the dLN but also within the tumor and tumor-associated lymphoid structures (such as TLSs). These insights affect the perspective on shaping the goals for the most effective T-cell responses (quality, trajectory, persistence may outweigh quantity). Similarly, these considerations should influence the understanding of T-cell immunity-related toxicity.
The challenges in developing immunotherapies reflect the complexity of human immunity, particularly a series of mechanisms that create rate-limiting steps at each consecutive stage of the CI cycle. This consideration goes beyond the presence of permissive or restrictive immune types, encompassing immune type-agnostic characteristics, which can best be described as shared mechanisms of immune escape.This mechanism involves the participation of both intrinsic and extrinsic factors in cancer, such as the loss or downregulation of Class I, the absence of neoantigens, the accumulation of multiple immune checkpoints, an increase in suppressive cells within the TME, and the loss of appropriate cell numbers.The early disease environment can at least avoid some of these mechanisms; machine learning models provided by relevant biomarker data may help alleviate them or propose new therapeutic combinations. Regardless of the approach, the goal remains to take appropriate steps to ensure the continuous evolution of the tumor-immune cycle.
Reference:The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity. 2023 Oct 10;56(10):2188-2205. doi: 10.1016/j.immuni.2023.09.011.