
Tuberculosis (TB) is a respiratory infectious disease caused by Mycobacterium tuberculosis (Mtb), and was the leading cause of death from infectious diseases globally before the COVID-19 pandemic. Previously, the treatment of tuberculosis mainly relied on linezolid and macrolide antibiotics. Linezolid inhibits protein synthesis by targeting the 50S ribosome, but its toxicity has hindered its widespread use in TB treatment. Macrolides block protein biosynthesis by obstructing the polypeptide exit tunnel of the ribosome. However, macrolide antibiotics induce the Mtb erm37 gene, which encodes a methyltransferase that methylates the A2058 site of the Mtb ribosome, altering ribosome conformation and affecting its binding to the ribosome. Therefore, although these antibiotics are cornerstone drugs for treating many bacterial infections, they have been proven ineffective against tuberculosis. With the growing issue of antibiotic resistance, the number of cases of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) continues to increase annually. Thus, there is an urgent need for new anti-tuberculosis drug combinations with anti-Mtb activity.
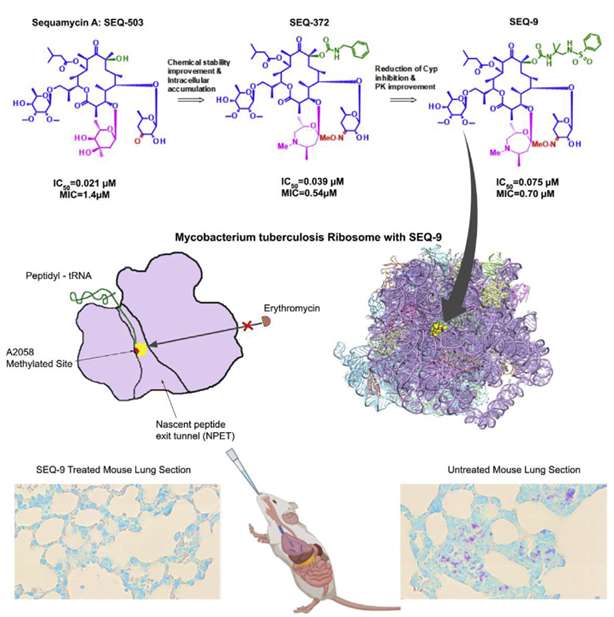
The team led by Sophie Lagrange from Sanofi, France, optimized and improved the natural product sequanamycin A to produce an advanced lead compound, SEQ-9. SEQ-9 was effective as a single drug in both acute and chronic tuberculosis models and demonstrated bactericidal activity when used in combination with other tuberculosis drugs in a mouse model of tuberculosis infection. These results support further research on this series as clinical candidates for tuberculosis and have the potential to become a new treatment option for drug-sensitive and drug-resistant tuberculosis. This research was recently published in the journal Cell (Zhang et al., 2023, Cell 186, 1013–1025).
Sequanamycin A (SEQ-503) was discovered in 1969 and is a 14-membered ring erythromycin family macrolide produced by Allokutzneria albata (a member of the Actinomycetales order and Pseudonocardiaceae family). Structurally, SEQ-503 differs from other 14-membered macrolides, featuring mycarose at the C3 position instead of cladinose, keto-allose at the C5 position instead of erythro deoxysamine, and maltose at the C13 position.
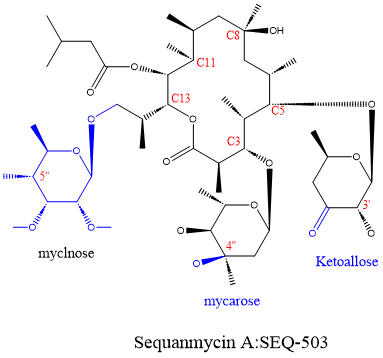
Figure 1. Molecular structure of sequanamycin A (SEQ-503)
Moreover, compared with most macrolide antibiotics, the minimum inhibitory concentration (MIC) of SEQ-503 against Mycobacterium tuberculosis is only 1.4 μM, demonstrating its potential as an anti-tuberculosis drug.
But this compound presents several challenges: First, SEQ-503 is unstable in acidic media: it has a half-life of less than 30 minutes in 50% acetonitrile/water at pH 2, indicating that it would rapidly degrade in the human stomach and thus is unsuitable as an oral drug. Second, the compound is metabolically unstable in human liver microsomes: 67% of the compound is metabolized within 20 minutes in vitro.
To overcome these limitations, researchers conducted medicinal chemistry modifications and studies on SEQ-503, discovering two spectinomycin derivatives, SEQ-569 and SEQ-977, with favorable inhibitory activity against the Mtb H37Rv strain (MICs of 0.57 and 0.13 mM, respectively). The overall structure of SEQ-977 binding to the ribosome is similar to that of SEQ-569, differing only in the substituent at the C8 position. Moreover, both SEQ-569 and SEQ-977 successfully bind to the T. thermophilus 70S ribosome.
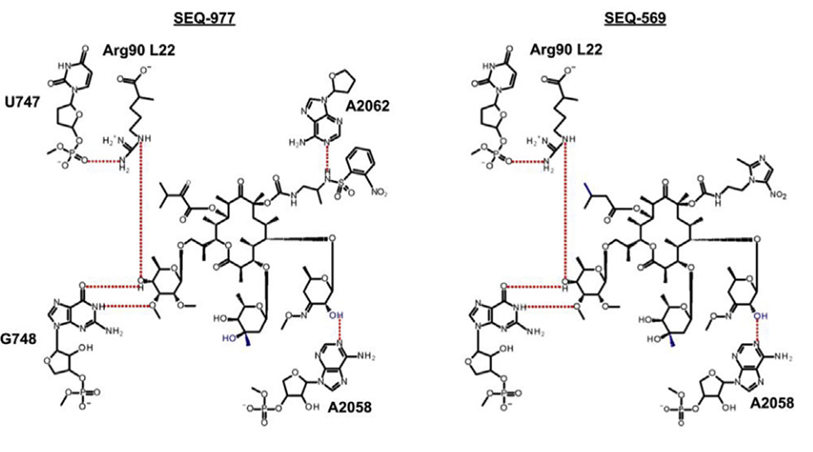
Figure 2. Molecular structures of SEQ-569 and SEQ-977
Compared with the classical erythromycin macrolides, the sequanamycin family has some additional hydrogen bonds that are crucial for the sequanamycin family of compounds. Taking SEQ-977 as an example, in addition to the carbamate group at the C8 position, the NH of the sulfonamide group also forms a hydrogen bond with the N1 of A2062. This interaction leads to a different orientation of A2062 in the co-crystal structure of SEQ-977 compared to SEQ-569, also explaining why SEQ-977 (IC50 = 0.034 μM, MIC = 0.13 μM) has higher potency than SEQ-569 (IC50 = 0.054 μM, MIC = 0.57 μM).
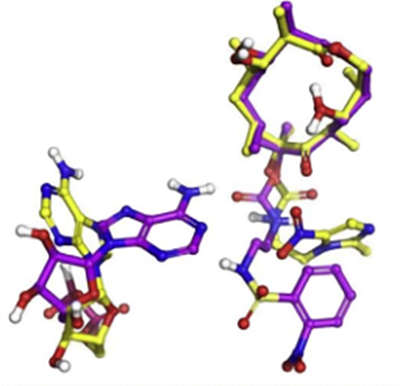
Figure 3. Relative positions of A2062 in the presence of carbamate at the C8 position of SEQ-569 (yellow) and SEQ-977 (purple)
The research team subsequently optimized the ketopentose modification at C5 and the carbamate group at C8, generating new compounds such as SEQ-416, which exhibited better anti-tuberculosis activity and chemical stability but showed limited efficacy in a mouse tuberculosis model. The researchers speculated that these results might be related to the limited penetration and accumulation in macrophages.
Mycarose was further modified to a 7-membered 1,4-oxazepane with a substituted amine, synthesizing SEQ-372, which has weak basicity (pKa = 8.2) and better lipophilicity (logD = 4.93). These physicochemical properties can improve the intracellular accumulation of the compound. Compared with its neutral analog SEQ-416 (IC50 = 0.044 μM, MIC = 0.15 μM), SEQ-372 (IC50 = 0.039 μM, MIC = 0.54 μM) showed slightly lower antibacterial efficacy under standard MIC assay conditions but exhibited a 20-fold increase in accumulation within macrophages and approximately a 10-fold increase in anti-Mtb potency inside macrophages.
SEQ-372 is the first compound in the series that has demonstrated dose-dependent efficacy in an acute TB infection mouse model, outperforming clarithromycin at the same dose and under the same assay conditions. However, the property of SEQ-372 to inhibit cytochrome 3A4 limits its use as a combination therapy drug.
Further optimization of the carbamate moiety at C8 led to the synthesis of SEQ-9 (IC50 = 0.065 μM), which exhibited ideal absorption, distribution, metabolism, and excretion (ADME) properties and lacked cytochrome 3A4 inhibitory activity. The half-life of this compound under acidic conditions (pH 2.0, 50% acetonitrile/water) increased from 30 minutes for SEQ-503 to 48 hours. Moreover, due to its lower metabolic burden in human, mouse, and rat microsomes, the area under the plasma concentration-time curve (AUC) of SEQ-9 was 6 times higher than that of SEQ-372 when administered orally to mice at the same dose (30 mg/kg). Additionally, the lung-to-plasma exposure ratio was 19, indicating enrichment in lung tissue. Researchers also found that SEQ-9 demonstrated a more suitable safety margin in in vitro cytotoxicity assays using HepG2 cells (IC50 = 10 μM) and primary human hepatocytes (IC50 = 26 μM).
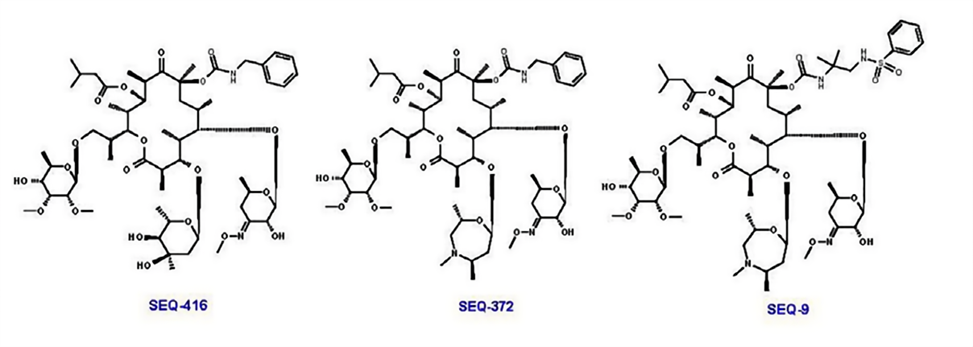
Figure 4. Molecular structures of SEQ-416, SEQ-372, and SEQ-9
Relevant experiments have demonstrated that SEQ-9 exhibits potent targeting activity against Mtb ribosomes and has a reasonable safety margin in vitro. To evaluate the resistance frequency to SEQ-9 and confirm its targeting activity, researchers isolated spontaneous Mtb mutants resistant to SEQ-9. It was found that the drug-resistant mutants selected by SEQ-9 showed higher resistance to sequanamycin antibiotics and clarithromycin while remaining sensitive to rifampicin and linezolid. To better understand the mechanism by which sequanamycin overcomes A2058 methylation resistance on the Mtb ribosome, researchers conducted cryo-electron microscopy studies to determine the structure of the A2058-methylated Mtb ribosome bound to SEQ-9 (PDB: 7KGB, EMDB: EMD22865) and compared it with the structure of the unmethylated Mtb ribosome bound to SEQ-9 (PDB: 7SFR, EMDB: EMD25100). Researchers achieved ribosomal methylation by culturing Mtb in medium with sub-inhibitory concentrations of erythromycin and measured the IC of the methylated ribosome compared to the unmethylated ribosome.50The change was confirmed by ribosome methylation. For the methylated Mtb ribosome, SEQ-9 still showed effective inhibition of protein synthesis (IC of methylated Mtb ribosome).50= 0.075 and 0.065 μM), indicating that the methylation at N6 of A2058 does not reduce the overall binding interaction between the Mtb ribosome and sequanamycin. The structure of the Staphylococcus aureus (S. aureus) ribosome suggests that macrolide resistance is caused by steric hindrance between the N6-methyl at A2058 and the dimethylamine of desosamine. This highlights the role of A2058 methylation in displacing water molecules, which are crucial for mediating antibiotic-ribosome binding.
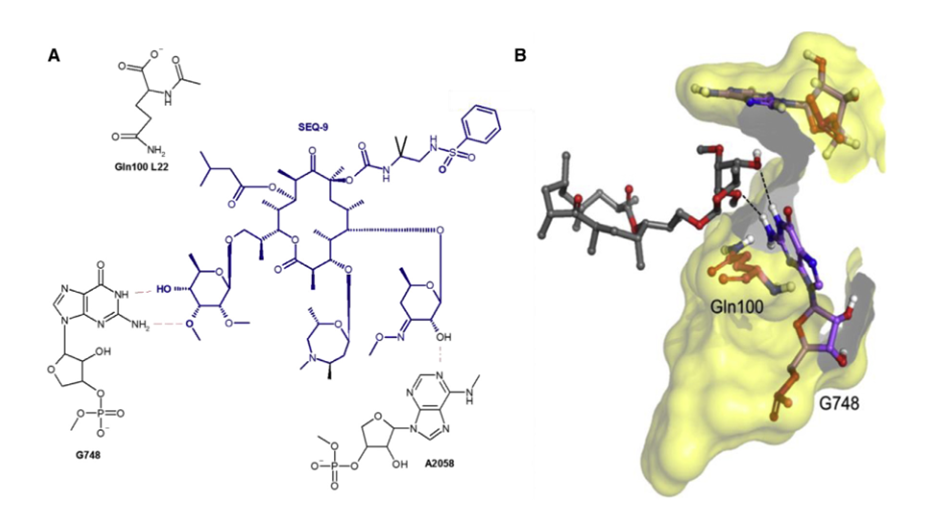
Figure 5. Interaction between SEQ-9 and Mtb Ribosome
In terms of efficacy, SEQ-9 demonstrated effective antibacterial activity against multiple Mtb strains and showed dose-dependent bactericidal effects in both acute and chronic TB mouse models. It also contributed significant bactericidal effects in combination therapies with Bedaquiline (B), Pretomanid (Pa), Linezolid (L), or Pyrazinamide (Z).
Summary:Researchers report the discovery of a series of macrolide compounds called sequanamycins, which exhibit excellent in vitro and in vivo activity against Mycobacterium tuberculosis (Mtb). Sequanamycin is a bacterial ribosome inhibitor that interacts with the ribosome in a manner similar to classical macrolide drugs such as erythromycin and clarithromycin, but with binding characteristics that enable them to overcome the inherent macrolide resistance of Mtb. The ribosome structure bound to the inhibitor was used to optimize sequanamycin, leading to the production of the advanced lead compound SEQ-9. SEQ-9 is effective as a single agent in both acute and chronic tuberculosis models and has shown bactericidal activity when combined with other tuberculosis drugs in a mouse model of Mtb infection. These results support further investigation of this series as clinical candidates for tuberculosis and have the potential to become a new treatment regimen for drug-sensitive and drug-resistant tuberculosis.
References
【1】Zhang J, Lair C, Roubert C, et al. Discovery of natural-product-derived sequanamycins as potent oral anti-tuberculosis agents. Cell. 2023,186,1013-1025