At the UK Biobank 2023 Scientific Conference held on December 13, 2023, there was a paper published on the medical preprint website medRxiv.Whole-genome sequencing of half-a-million UK Biobank participantsHas sparked widespread attention in academic circles.
This study conducted whole-genome sequencing on approximately 500,000 participants, including individuals from five ethnic groups, marking another milestone in human genetic exploration. Although this research has not yet been officially peer-reviewed and published, we can still get a sneak peek through this preprint.
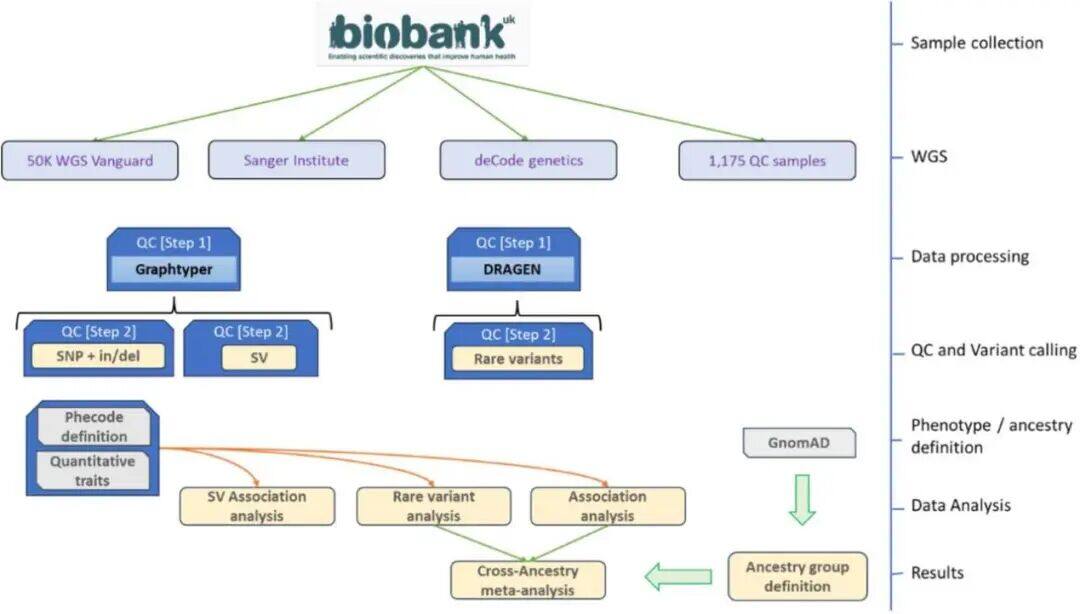
 With the continuous development of gene sequencing technology, our understanding of genes is also increasing. However, the more we understand, the more unknown areas are revealed. To this end, UK Biobank has launched a large-scale whole genome sequencing (WGS) project covering nearly 500,000 people, based on previous genotyping and whole exome sequencing (WES) studies. This is also the largest whole genome sequencing effort to date, encompassing five ethnic groups.The figure below shows the research framework:Researchers first collected patient samples through the UK Biobank, followed by WGS. Quality control was performed on the sequencing results using the GraphTyper and DRAGEN datasets, followed by variant calling for SNPs, in/dels, and structural variants (SVs). Subsequently, researchers defined phenotypes (binary and quantitative) related to SVs, SNPs, and gene levels (rare variant analysis) and drew conclusions. The five ethnic groups—NFE (non-Finnish Europeans), SAS (South Asians), AFR (Africans), ASJ (Ashkenazi Jews), and EAS (East Asians)—and the collective association effects were defined as cross-ancestry meta-analysis.
With the continuous development of gene sequencing technology, our understanding of genes is also increasing. However, the more we understand, the more unknown areas are revealed. To this end, UK Biobank has launched a large-scale whole genome sequencing (WGS) project covering nearly 500,000 people, based on previous genotyping and whole exome sequencing (WES) studies. This is also the largest whole genome sequencing effort to date, encompassing five ethnic groups.The figure below shows the research framework:Researchers first collected patient samples through the UK Biobank, followed by WGS. Quality control was performed on the sequencing results using the GraphTyper and DRAGEN datasets, followed by variant calling for SNPs, in/dels, and structural variants (SVs). Subsequently, researchers defined phenotypes (binary and quantitative) related to SVs, SNPs, and gene levels (rare variant analysis) and drew conclusions. The five ethnic groups—NFE (non-Finnish Europeans), SAS (South Asians), AFR (Africans), ASJ (Ashkenazi Jews), and EAS (East Asians)—and the collective association effects were defined as cross-ancestry meta-analysis.Sample Sources and Methods
The study utilized the Illumina NovaSeq™ 6000 sequencing platform to perform WGS on 490,640 participants from the UK Biobank. The average sequencing depth for the WGS was 32.5x, with duplicate samples used for quality control.
Subsequently, Illumina's DRAGEN Bio-IT Platform successfully processed a large number of SNP and indel variants (both within the high accuracy range), with SNP detection sensitivity at 99.77% and precision at 99.91%; indel sensitivity at 99.70% and precision at 99.83%.Finally, perform single-variant testing, multi-ancestry meta-analysis, rare variant aggregation analysis, and structural variant analysis on the WSG sequencing results, GraphTyper, and DRAGEN system’s variant calling results.Figure 1Description of Variant Types and Frequencies
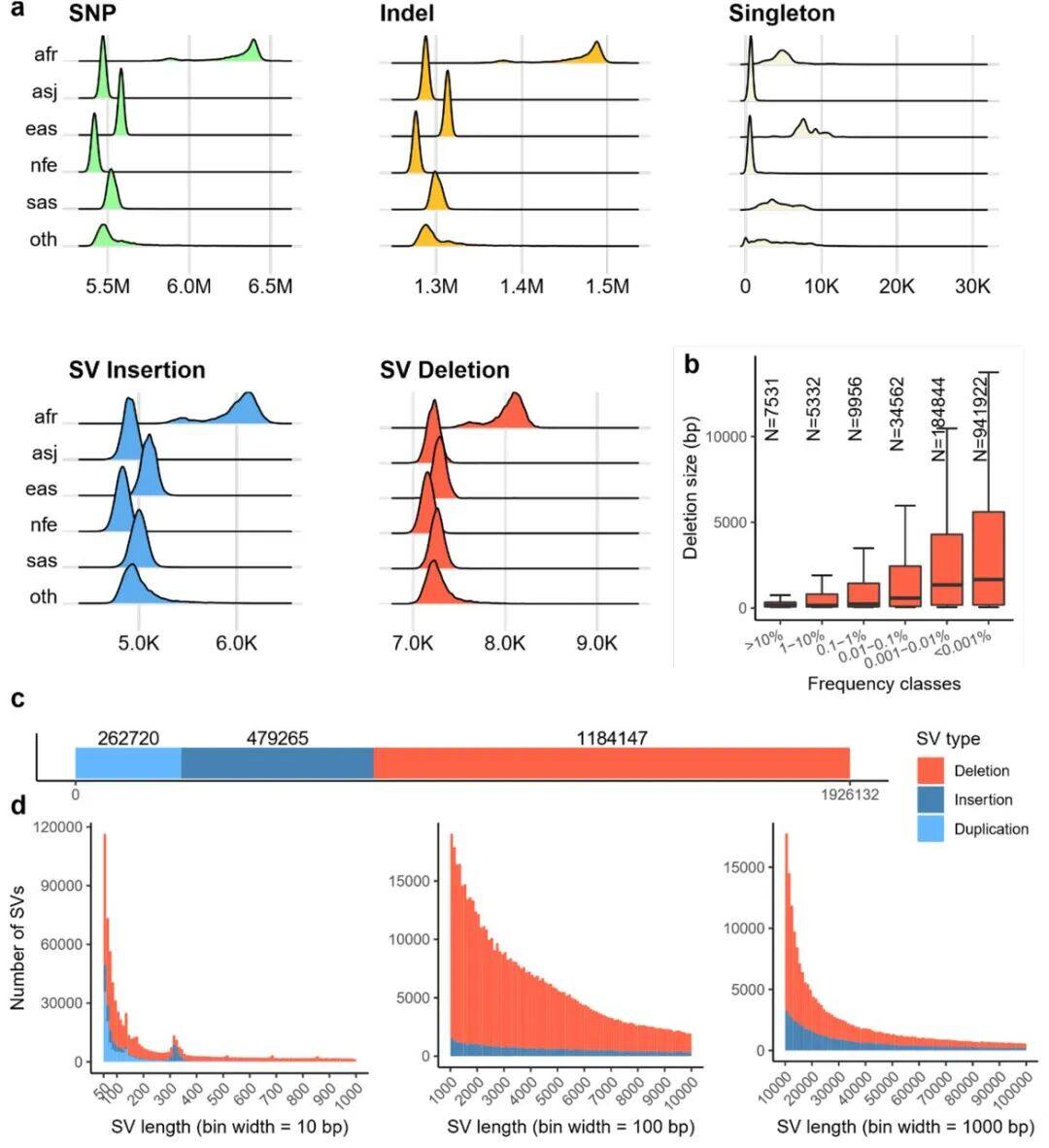
Figure 1 consists of multiple subfigures, involving different types and frequency distributions of genomic variations:
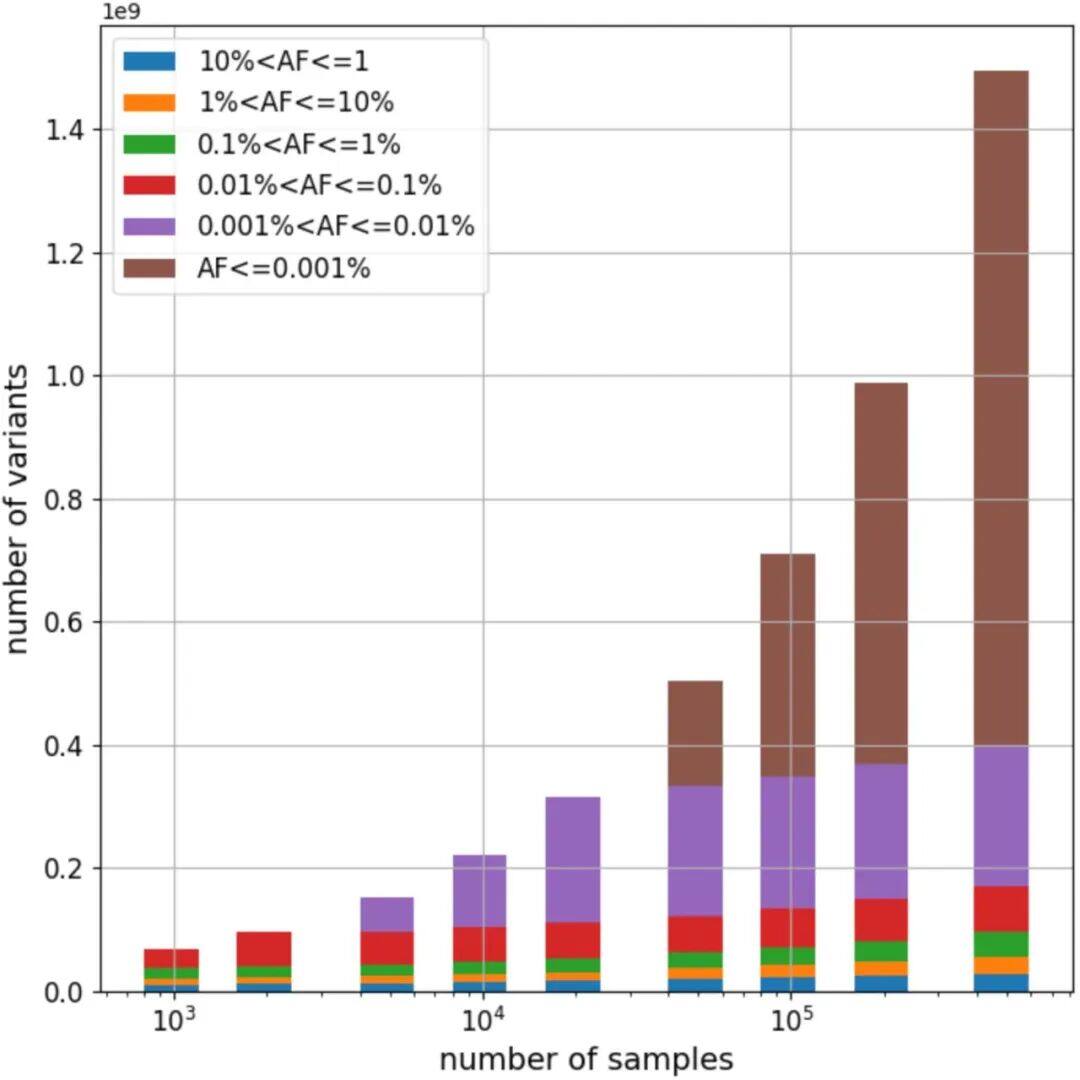
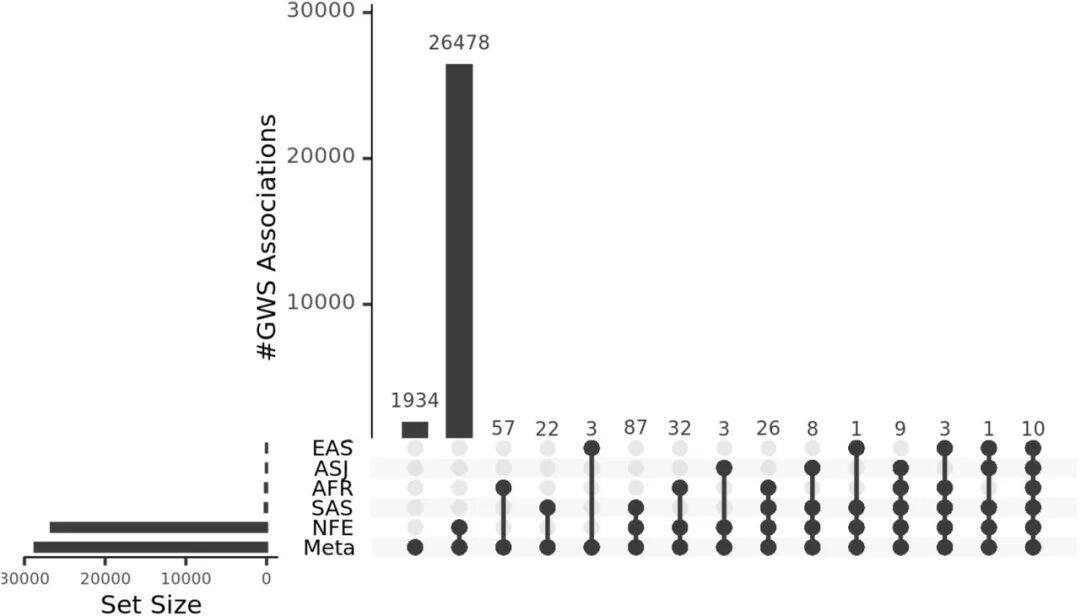
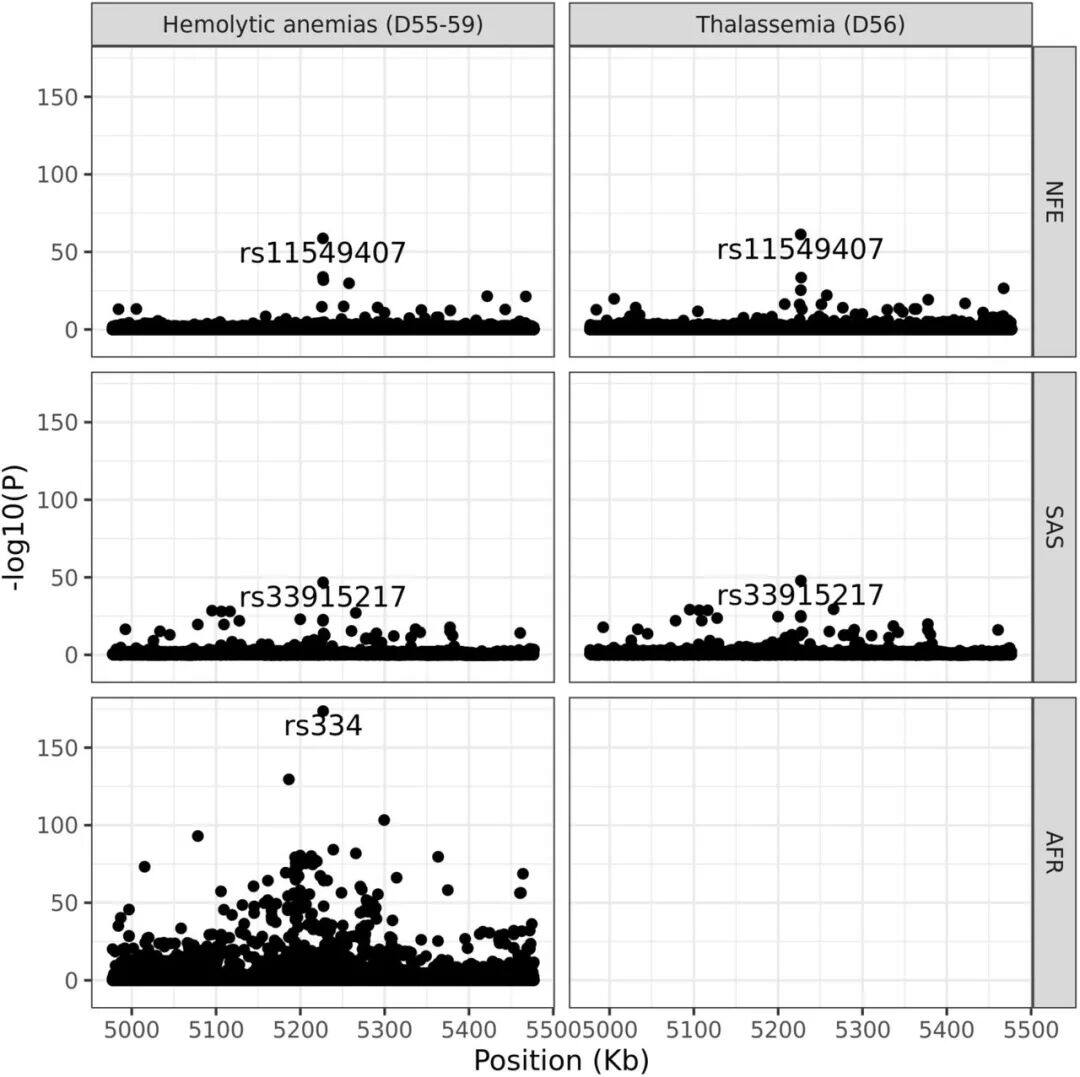
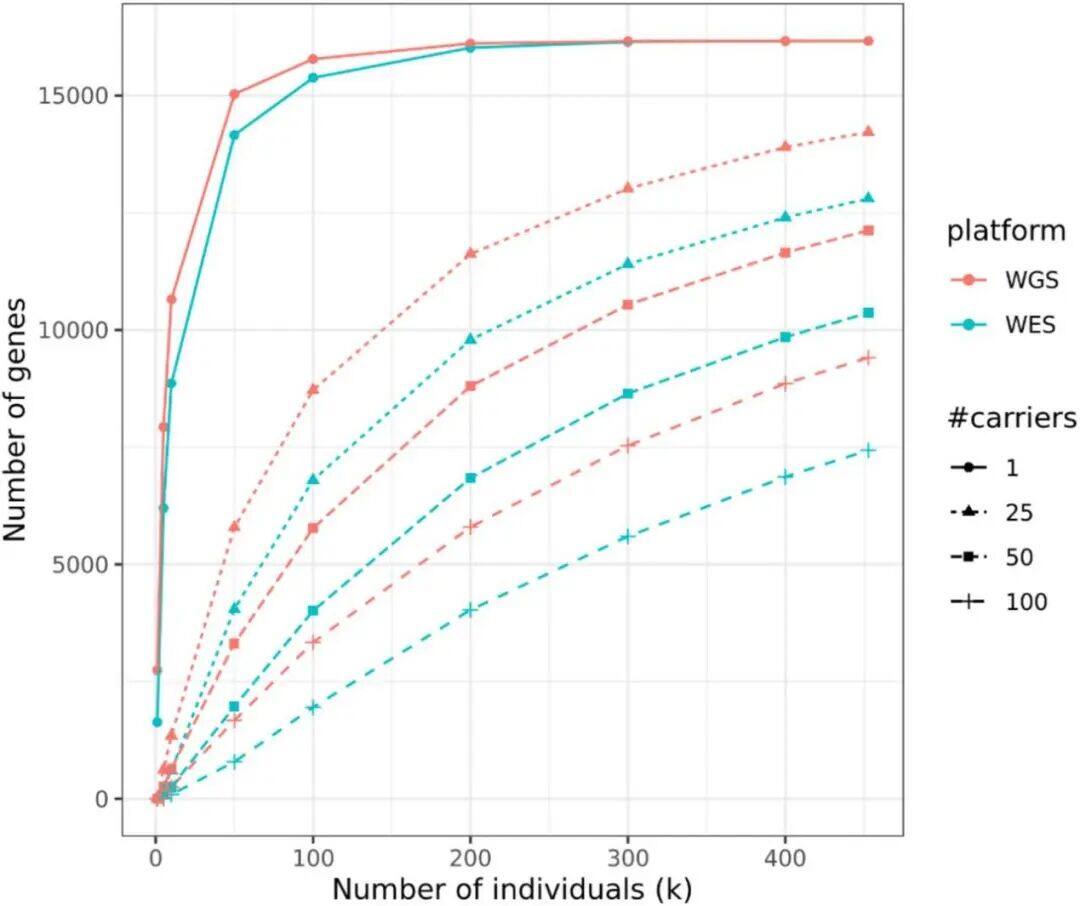
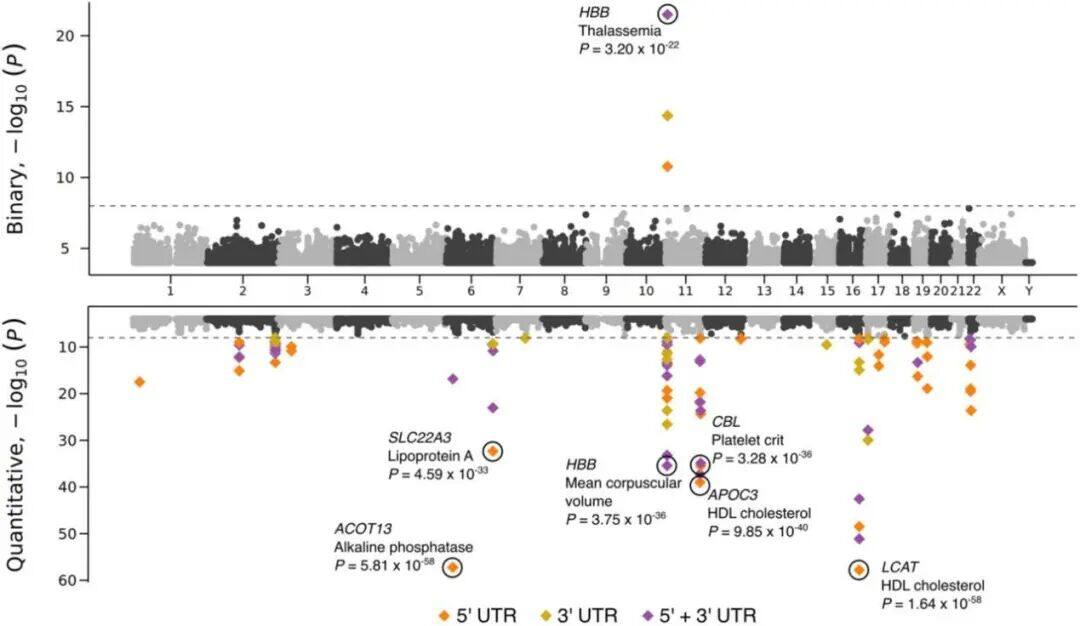
a) Density/number of each type of variant in different populations: This subplot shows the number of variants identified per individual across five populations with different ancestral backgrounds. Specifically, the types of variants include SNPs (single nucleotide polymorphisms), indels (insertions or deletions), singleton SNPs/indels (SNPs/indels that occur only once), insertions/duplications in structural variants, and deletions.b) Length distribution of structural variations (SVs): This subgraph focuses on the size distribution of SVs, with different frequencies of variations distinguished by color codes. The red area represents the 25th to 75th percentile values of variation sizes, the straight line indicates the median, and the top horizontal line shows the 95% confidence interval. The length distribution reveals a tendency toward shorter SVs.c) Number of different types of SVs: This shows the number of SVs categorized by variant type (duplication, insertion, deletion).d) Size distribution of insertions and deletions: This subgraph specifies the size range of insertions and deletions, categorizing variations from 50bp to 1,000bp, 10,000bp, and even up to 100,000bp.Figure 2Analysis of Increased Variability in Sample SizeFigure 2 depicts how the number of variants across different allele frequency ranges is revealed through varying sample sizes (from 1,000 to 490,541). Specifically, the x-axis represents the number of samples in the random downsampling analysis, and the y-axis represents the number of identified variants.As shown in Figure 2: For common variants (e.g., >1% frequency), even with an increase in sample size, we only observed a slight increase in the number of variants. For rare variants (e.g., <=0.001% frequency), the number of variants increased significantly as the sample size grew, and no plateau was observed even at the largest sample sizes, indicating that continued large-scale sequencing efforts are valuable for discovering new and high-impact rare variants.Figure 3Disruption Map of GWS (Genome-Wide Significance) Associations Across AncestriesFigure 3 is an UpSet interaction diagram, commonly used to display the distribution of intersections and unique elements among multiple sets. It shows the distribution of genome-wide significant (GWS) associations, including meta-analyses across different ancestries (ethnic groups) and associations within specific ancestries. The x-axis labels are sorted by the number of GWS associations: meta-analysis, NFE (non-Finnish Europeans), SAS (South Asians), AFR (Africans), ASJ (Ashkenazi Jews), and EAS (East Asians).According to Figure 3, the largest number of GWS associations were found in non-Finnish European populations, followed closely by meta-analysis, indicating that certain gene-trait associations span multiple ethnic groups. The relatively small number of GWS discoveries in non-European populations also suggests there is room for further exploration of these population-specific genetic associations.Figure 4Association between the HBB-HBE1 locus and anemia-related diseasesThe left half of Figure 4 shows the association between HBB-HBE1 and hemolytic anemia (D55-59) in non-Finnish Europeans (NFE), Africans (AFR), and South Asians (SAS), while the right half displays the association between HBB-HBE1 and thalassemia (Thalassaemia, D56) in NFE, AFR, and SAS populations.Figure 4 shows the distribution of association signals for a specific trait— anemia — across different populations. The figure highlights certain single nucleotide polymorphisms (SNPs), such as rs334. This variant is a common cause of SCD (sickle cell disease) and is associated with a missense mutation in the HBB gene. These associations reveal the link between genetic variations and disease phenotypes, as well as how these associations exhibit different epidemiological characteristics across ethnic groups. rs334 is more common in African populations but rarer in non-Finnish Europeans and South Asians. The findings uncover traces of natural selection based on population-specific gene frequencies; for example, the protective effect of certain loci against malaria may result in higher variant frequencies in specific groups. rs334 is also associated with other biochemical indicators (such as creatinine and urea), consistent with the clinical manifestations of sickle cell disease. Similarly, association studies on thalassemia-related traits provide further evidence of the genetic influence on disease phenotypes.Figure 5Number of people carrying harmful pLoF/P/LP variant genesFigure 5 describes the number of genes found to carry deleterious variants in whole-genome sequencing (WGS) and whole-exome sequencing (WES) datasets. pLoF refers to predicted loss-of-function variants, and P/LP refers to clinically recognized pathogenic/likely pathogenic variants. The X-axis shows the number of participants, and the Y-axis represents the number of autosomal genes with at least a specific number (1, 25, 50, 100) of carriers.Figure 5 shows that the number of genes detected by WGS with at least 100 harmful variants exceeds the number of genes in the WES dataset. Figure 5 highlights the comparison between WGS and WES datasets in revealing harmful variants, demonstrating the importance of WGS in discovering and understanding rare and potentially harmful genetic variants that contribute to the risk of specific genetic disorders. Through WGS, we are able to identify harmful variants within a broader genomic range that may not have been detected in WES, which is crucial for etiological research and potential targeted therapies.Figure 6 Association between UTR genotype and phenotypeFigure 6 is a collapse analysis diagram showing the combined analysis of rare UTR variants and phenotype associations. The upper part of the image displays the association analysis results for binary traits, while the lower part shows the association analysis results for quantitative traits. Different colors represent different UTR models (5’ UTR, 3’ UTR, and both combined), and significant association signals with P-values < 1e-30 are annotated in the figure.Figure 6 illustrates that rare UTR variants do have significant associations with certain biological traits and disease phenotypes. In some cases, the joint analysis of pure UTR variants with specific phenotypes can lead to new statistically significant associations. This suggests that UTR variants may play an important role in post-transcriptional regulation and related disease processes, which might have been overlooked in previous studies based on WES technology. For traits that have signals only in protein-coding regions while UTR variants were not considered, this finding implies that exploration of rare variants and non-coding regions should be expanded to better understand disease mechanisms.
In this study, WGS detected approximately 150 million variants, including single nucleotide polymorphisms (SNPs), insertions and deletions (indels), and structural variations (SVs), which is more than 18.8 times and 40 times higher than observed through genotype imputation and WES in human variations. Compared to the variants discovered solely through WES, WGS covers more protein-coding regions as well as variants in the 5’ and 3’ untranslated regions (UTR) sequences. Large-scale WGS data brings an expanded potential for understanding the role of rare non-coding variants in health and disease, such as:WGS data shows that the number of variations captured in all annotation categories exceeds that of WES. Structural variations (SVs), although fewer in number than SNPs and indels, affect a comparable average number of base pairs as SNPs and indels. Through multi-ancestry meta-analysis, it was found that a reduction in cytochrome C-reactive protein is associated with the APOE gene mutation rs429358-C, which is commonly believed to increase the risk of Alzheimer’s disease. The detection of structural variations can reveal new associations between genes and traits; for example, SVs in the PCSK9 gene are significantly associated with non-HDL cholesterol levels.This study enhances our understanding of human genetic variation and its impact on disease occurrence, and will lay the groundwork for the discovery of new diagnostic methods, more effective and safer therapies, as well as the development of precision medicine strategies that may improve global health. Additionally, this research establishes a foundation for exploring the role of "selective forces" in shaping human genetic diversity through pathogenic and protective variants.The whole genome sequencing (WGS) carried out by the UK Biobank project used the Illumina NovaSeq™ 6000 sequencing platform, with sequencing conducted at deCODE Genetics in Iceland and the Wellcome Sanger Institute in the UK. Dr. Mark Effingham, Deputy Chief Executive Officer of UK Biobank, also stated: "Whole genome sequencing on such a large scale has extremely high requirements for the accuracy and sensitivity of the technology."Illumina Became the Preferred Technical Partner of UK Biobank at That Time.Moreover, it is worth noting that the project's analysis also utilized the renowned Illumina DRAGEN Germline analysis pipeline, known for its efficiency and accuracy. This analytical process aligns with those used in other large-scale population genomics initiatives to facilitate future cross-analysis of data. These large-scale population genomic initiatives include Singapore’s National Precision Medicine program PRECISE, the UK’s Genomics England 100,000 Genomes Project, the All of Us research program—a million-person cohort study supported by the U.S. National Institutes of Health—and the Alliance for Genomic Discovery led by Nashville Biosciences.The original text link of this paper:https://www.medrxiv.org/content/10.1101/2023.12.06.23299426v1