Disclaimer: Due to limited proficiency, errors are inevitable, and some information may not be the most up-to-date. Comments are welcome to point out any issues. This article is only an introduction to pharmaceuticals related to medical health, not a recommendation of treatment options (if involved); this article does not constitute any investment advice.
As the global benchmark for pharmaceutical regulation, the U.S. Food and Drug Administration (FDA) holds the lifeline of innovative drug development companies. Its decision to approve or reject a drug's market entry directly determines the fate of many clinical-stage innovative drug companies, ensuring the safety and efficacy of products. At the start of the new year, which new drugs have been rejected by the FDA? Let’s take a look.
CLDN18.2 Monoclonal Antibody Zolbetuximab: CLDN18.2-positive Locally Advanced Unresectable or Metastatic HER2-negative Gastric Cancer or Gastroesophageal Junction (GEJ) Adenocarcinoma
On January 9, 2024, Astellas announced that it had received feedback from the FDA regarding its CLDN18.2 monoclonal antibody.zolbetuximabComplete Response Letter (CRL) for Marketing ApplicationTemporarily rejected for marketing approval as a first-line treatment for locally advanced unresectable or metastatic gastric or gastroesophageal junction (G/GEJ) adenocarcinoma that is Claudin18.2-positive and HER2-negative.

In the CRL letter,FDA Points Out Unresolved Deficiencies at Third-Party Manufacturing Plant for Zolbetuximab, indicating that the drug cannot be approved for marketing before the original PDUFA date. Notably, the FDA did not raise any questions regarding the clinical data of zolbetuximab, nor did it request additional clinical trials. Astellas stated that these manufacturing deficiencies do not pose any safety or efficacy concerns. The company is working closely with the FDA and third-party manufacturers to resolve the issue as soon as possible.Astellas Focus Areas Pipeline Layout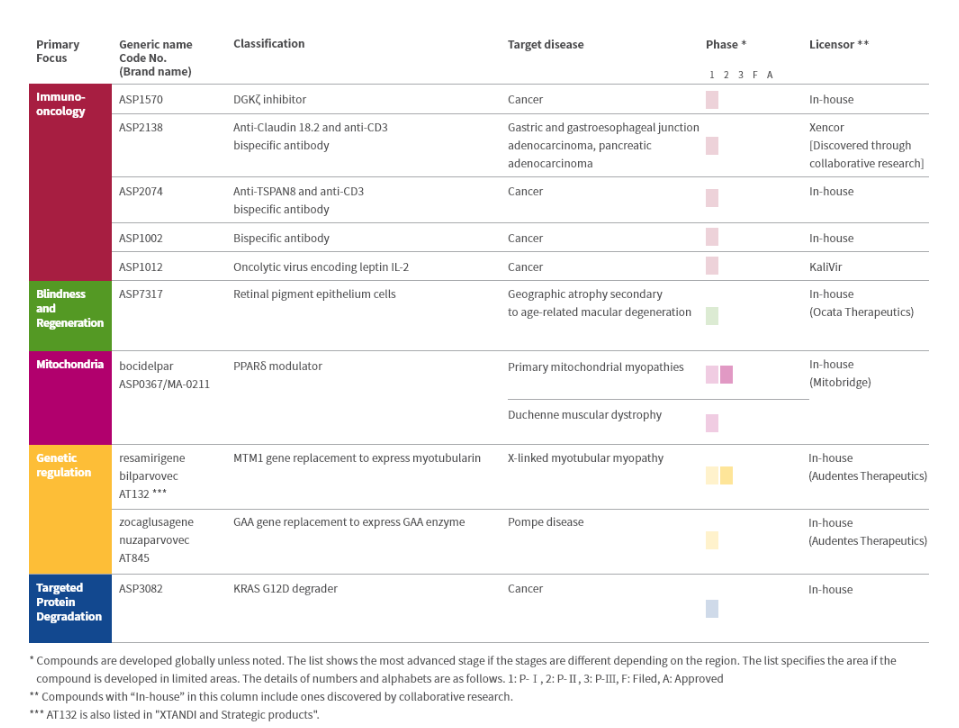
Dihydroergotamine Nasal Powder Product STS101:Acute Migraine
January 18, 2024FDA Rejects Approval of Satsuma Pharmaceuticals’ Dihydroergotamine Nasal Powder Product STS101 for Acute Migraine TreatmentSTS101 is a reformulated version of the nasal-administered migraine medication dihydroergotamine mesylate (DHE), delivered using Satsuma's proprietary device, which is only the size of a lipstick, making it convenient to carry and use.The complete response letter (CRL) pointed out the issues related to the chemistry, manufacturing, and controls (CMC) of STS101.CRL did not indicate any safety issues with STS101 and did not request additional studies.Satsuma to Discuss Refusal with FDA for Resubmission of Marketing Application.The marketing application of Satsuma is based on the Phase III clinical trials ASCEND study and SUMMIT study.In the ASCEND study, more than 10,500 doses of STS101 were administered to 446 enrolled patients within 18 months. Two hours post-treatment, STS101 provided pain freedom in 34.2% of all treated migraine attacks, freedom from the most bothersome symptoms in 53.4% of patients, and eliminated the need for rescue medication in 94% of treated attacks.However, the SUMMIT study did not meet its primary efficacy endpoint, as there was no significant difference between STS101 and placebo in terms of being pain-free and having the most bothersome symptoms resolved two hours post-treatment.In terms of safety, no serious adverse events were observed with STS101, and most toxicities were mild and transient.Intranasal Scopolamine Gel:Nausea and Vomiting Caused by Motion Sickness
January 30,Defender PharmaceuticalsAnnounce FDAIssued a Complete Response Letter (CRL),Rejected the company's application forNDA for Intranasal Scopolamine Gel (DPI-386 Nasal Gel) to Prevent Nausea and Vomiting Caused by Motion Sickness。The company did not explain the reason for the regulatory authority's rejection of the drug.
DPI-386 Gel Was Evaluated in a Phase 3, Double-Blind, Placebo-Controlled Study (NCT05548270) Involving Approximately 500 Adults Exposed to Motion During Sea Travel. Participants Were Randomly Assigned to Self-Administer Either Scopolamine Nasal Gel (Containing 0.2 mg of Scopolamine HBr Per 0.12 g of Gel) or a Placebo. The Study Demonstrated That a Significantly Higher Proportion of Patients in the Intranasal Scopolamine Group Reported No Vomiting and Did Not Request Rescue Medication (Primary Endpoint) Compared to the Placebo Group.Defender Pharmaceuticals, Inc.Barry I. Feinberg, M.D., President and Chief Executive Officer, said:“Defender Pharmaceuticals still believes that intranasal scopolamine is a safe and effective therapy for the prevention of motion sickness, and we will work closely with the FDA to ensure that we can bring this innovative new product to market."DefenderWill continue to seek innovative uses for scopolamine and plans to study its potential in a range of indications including traumatic brain injury, postoperative nausea and vomiting.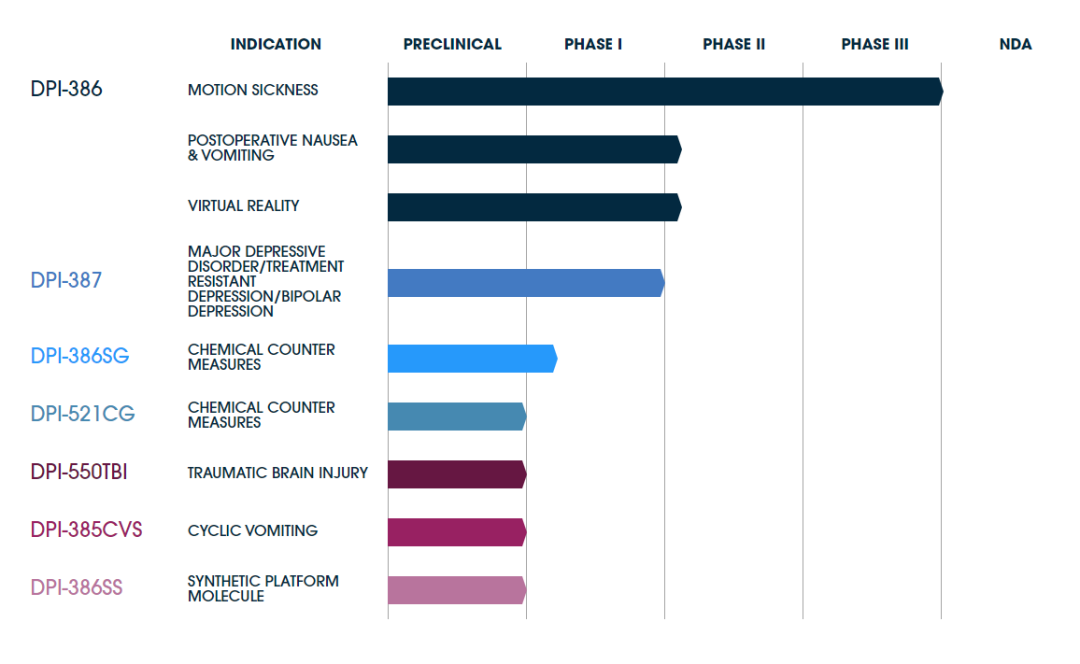
Roluperidone:Negative Symptoms of Schizophrenia
On February 27, Minerva Neurosciences, Inc. announced that it had received a Complete Response Letter (CRL) from the FDA regarding the New Drug Application (NDA) for Roluperidone intended for the treatment of negative symptoms of schizophrenia.Rejected its new schizophreniaMedicine.Affected by this news, Minerva's stock price plummeted by 59%.In CRL, the FDA pointed out the following four reasons for rejection:Although the MIN-101C03 study demonstrated statistical significance on the primary efficacy endpoint, it alone is insufficient to establish substantial evidence of effectiveness; the submitted NDA lacks data regarding concomitant use of antipsychotic medications;The NDA submission lacks the necessary data to demonstrate that Roluperidone has a clinically meaningful effect on improving negative symptoms of schizophrenia; among the safety data submitted, there are insufficient patients who have taken the proposed dose (64mg) of Roluperidone for at least 12 months.
To address these deficiencies, the FDA required Minerva to submit at least one additional positive, adequate, and well-controlled study to support the safety and efficacy of Roluperidone in treating negative symptoms.Minerva must also provide additional data to demonstrate the safety and efficacy of Roluperidone in combination with other antipsychotic drugs, further supporting Roluperidone's efficacy and proving the long-term safety of the proposed dose.
On February 27, Theratechnologies (NASDAQ: THTX) announced that the U.S. Food and Drug Administration (FDA) had issued a Refusal to File (RTF) letter regarding the company's supplemental Biologics License Application (sBLA) for the intramuscular (IM) administration method of the maintenance dose of Trogarzo® (ibalizumab-uiyk).After an initial review, the FDA determined that the sBLA was not sufficiently complete to permit a substantive review. The RTF letter indicated that the sBLA did not include the data necessary to establish a pharmacokinetic bridge between the Trogarzo® IM administration route and the intravenous infusion route.
Trogarzo® (ibalizumab-uiyk) is a long-acting, CD4-directed, post-attachment HIV-1 inhibitor. In the United States, Trogarzo® is used in combination with other antiretroviral drugs for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in heavily treatment-experienced adults with multidrug-resistant HIV-1 infection who are failing current antiretroviral therapy. Trogarzo® is not approved in Canada.
According to incomplete statistics, in the past year of 2023, 31 new drugs were rejected by the FDA, involving tumors, heart diseases, respiratory system diseases, skin diseases, and so on.The reasons for rejection are overwhelmingly focused on questions regarding the product's efficacy and safety, including issues such as questionable clinical benefits and limited clinical data.In addition to clinical issues, CMC (Chemistry, Manufacturing, and Controls) issues have also begun to emerge as another significant factor for FDA refusal of approval., thisThis indicates that regulatory agencies are imposing increasingly stringent CMC requirements, and companies cannot afford to overlook CMC issues.Reference: Official websites of various companies





