GLPG3970It is an SIK2/3 inhibitor developed by Galapagos. The development of this drug began in 2019 under a collaboration agreement between the two parties, involving an amount as high as 5.1 billion US dollars. It was once considered a potential treatment for various autoimmune diseases. In 2021, the cooperation between Galapagos and Gilead faced a crisis. In a Phase 2a clinical trial targeting rheumatoid arthritis and ulcerative colitis, the SIK2/3 inhibitor GLPG3970 failed to meet the endpoint. On March 30, 2024, the Journal of Medicinal Chemistry published online part of the research and development data of Galapagos' clinical candidate compound GLPG3970 for reference. Salt-Inducible Kinases (SIKs) belong to the AMPK family and have three subtypes: SIK1, SIK2, and SIK3. SIKs are widely expressed in tissues and possess similar kinase domains, making the development of selective SIK kinase inhibitors highly challenging. The mRNA encoding SIK1 is regulated by various stimuli, including high dietary salt intake, adrenocorticotropic hormone signaling, glucagon signaling, circadian rhythms, and more. In contrast, the expression of SIK2 and SIK3 is constitutive in tissues, and little is currently known about ligands that increase or decrease their expression levels. The activity of SIK is induced by LKB1 phosphorylation of its activation loop and inhibited by cAMP/PKA-dependent phosphorylation, which enhances binding to 14-3-3 proteins. Key substrates of SIK include CREB-regulated transcriptional coactivators (CRTCs) and histone deacetylases (HDACs). SIKs playCRTCsPhosphorylation of HDAC family members induces their binding to 14-3-3 proteins and retention in the cytoplasm. Dephosphorylation and subsequent translocation of CRTCs and HDACs to the nucleus, either through cAMP/PKA pathway-dependent phosphorylation or direct pharmacological inhibition of SIK, respectively promotes or inhibits gene transcription. The impact of SIK inhibition on immune responses has been explored in both innate and adaptive immune cells. Studies using pan-SIK inhibitors in yeast polysaccharide-stimulated mouse bone marrow-derived dendritic cells (BMDCs) have shown an increase in the production of the immunomodulatory cytokine interleukin-10 (IL-10), associated with a reduction in the secretion of pro-inflammatory cytokines such as tumor necrosis factor α (TNFα), IL-12, IL-1β, and IL-6. Additionally, in human macrophages stimulated by lipopolysaccharide (LPS) or IL-1β, SIK inhibition significantly reduces the phosphorylation of CRTC3 and HDAC4 substrates, thereby modulating pro-inflammatory and anti-inflammatory cytokines. Inhibition of SIK during mouse macrophage differentiation induces a phenotypic shift towards regulatory macrophages, characterized by a reduced ability to produce pro-inflammatory cytokines upon LPS stimulation, coupled with enhanced IL-10 production. SIK inhibitors represent a promising new approach for treating inflammatory diseases, particularly those characterized by immune system imbalance, such as IBD.
Salt-Inducible Kinases (SIKs) belong to the AMPK family and have three subtypes: SIK1, SIK2, and SIK3. SIKs are widely expressed in tissues and possess similar kinase domains, making the development of selective SIK kinase inhibitors highly challenging. The mRNA encoding SIK1 is regulated by various stimuli, including high dietary salt intake, adrenocorticotropic hormone signaling, glucagon signaling, circadian rhythms, and more. In contrast, the expression of SIK2 and SIK3 is constitutive in tissues, and little is currently known about ligands that increase or decrease their expression levels. The activity of SIK is induced by LKB1 phosphorylation of its activation loop and inhibited by cAMP/PKA-dependent phosphorylation, which enhances binding to 14-3-3 proteins. Key substrates of SIK include CREB-regulated transcriptional coactivators (CRTCs) and histone deacetylases (HDACs). SIKs playCRTCsPhosphorylation of HDAC family members induces their binding to 14-3-3 proteins and retention in the cytoplasm. Dephosphorylation and subsequent translocation of CRTCs and HDACs to the nucleus, either through cAMP/PKA pathway-dependent phosphorylation or direct pharmacological inhibition of SIK, respectively promotes or inhibits gene transcription. The impact of SIK inhibition on immune responses has been explored in both innate and adaptive immune cells. Studies using pan-SIK inhibitors in yeast polysaccharide-stimulated mouse bone marrow-derived dendritic cells (BMDCs) have shown an increase in the production of the immunomodulatory cytokine interleukin-10 (IL-10), associated with a reduction in the secretion of pro-inflammatory cytokines such as tumor necrosis factor α (TNFα), IL-12, IL-1β, and IL-6. Additionally, in human macrophages stimulated by lipopolysaccharide (LPS) or IL-1β, SIK inhibition significantly reduces the phosphorylation of CRTC3 and HDAC4 substrates, thereby modulating pro-inflammatory and anti-inflammatory cytokines. Inhibition of SIK during mouse macrophage differentiation induces a phenotypic shift towards regulatory macrophages, characterized by a reduced ability to produce pro-inflammatory cytokines upon LPS stimulation, coupled with enhanced IL-10 production. SIK inhibitors represent a promising new approach for treating inflammatory diseases, particularly those characterized by immune system imbalance, such as IBD.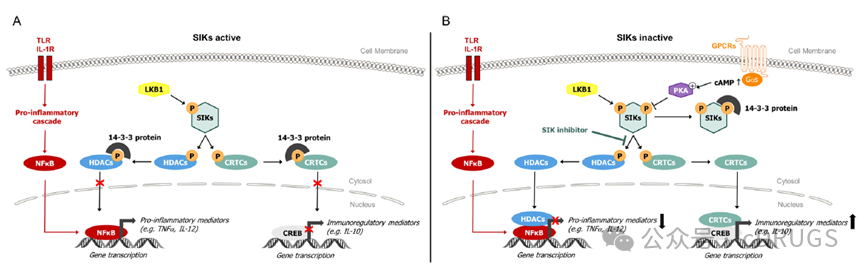
SIK Signaling Pathway Diagram
Enzymatic activity of endogenous SIK isoforms in mouse fetal liver-derived macrophages (FLDMs) and bone marrow-derived macrophages (BMDMs) was determined by immunoprecipitation. The enzymatic activity of SIK2 was higher than that of SIK3, while the contribution of SIK1 was the lowest. SIK2 and SIK3 were also found to be essential for the secretion of inflammatory mediators in IL-33-stimulated mouse bone marrow-derived mast cells (BMMCs). Exploration of the role of SIK isoforms in mouse T-cell development revealed that SIK2 and SIK3 are the predominant functional isoforms in T cells. Combined knockout of SIK2 and SIK3 resulted in a severe block in thymic T-cell development, consistent with defects in positive or negative selection of double-positive thymocytes. Therefore, dual inhibition of SIK2/SIK3 may offer better safety compared to pan-SIK inhibition, with reduced impact on the cardiovascular system while retaining the desired activity on the immune system.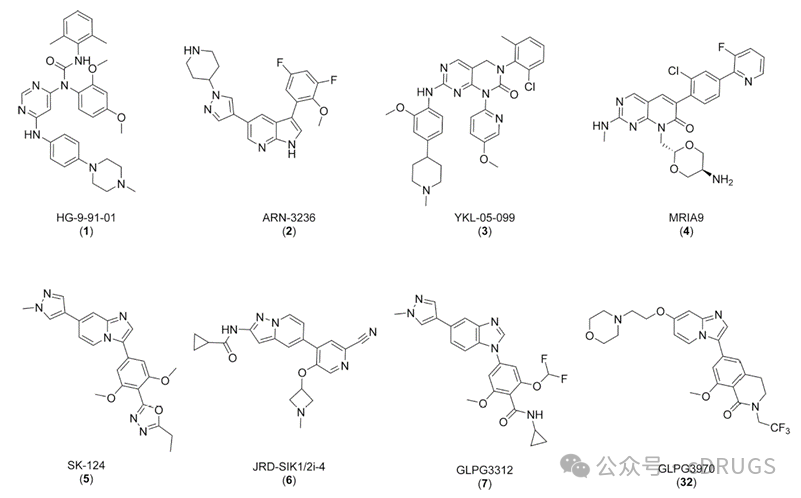 There is an increasing number of reports on small-molecule SIK inhibitors. Enhancing selectivity for other kinases and selectivity among SIK subtypes is a key goal in the development of new SIK inhibitors, thereby providing tools for studying the pharmacology of SIK. Based on the previously disclosed potent and selective pan-SIK inhibitor GLPG3312 and the SIK3 crystal structure, the authors, while studying GLPG3312, utilized the SIK3 crystal structure to explore the possibility of achieving effective dual inhibition of SIK2 and SIK3 through selective inhibition of SIK1, aiming to avoid the aforementioned SIK1-related side effects.
There is an increasing number of reports on small-molecule SIK inhibitors. Enhancing selectivity for other kinases and selectivity among SIK subtypes is a key goal in the development of new SIK inhibitors, thereby providing tools for studying the pharmacology of SIK. Based on the previously disclosed potent and selective pan-SIK inhibitor GLPG3312 and the SIK3 crystal structure, the authors, while studying GLPG3312, utilized the SIK3 crystal structure to explore the possibility of achieving effective dual inhibition of SIK2 and SIK3 through selective inhibition of SIK1, aiming to avoid the aforementioned SIK1-related side effects.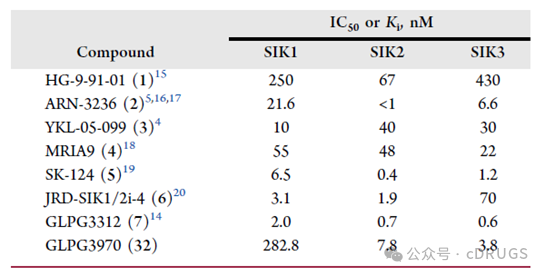 GLPG3970 is a dual SIK2/SIK3 inhibitor that has been evaluated in clinical trials for autoimmune and inflammatory diseases. The authors improved the selectivity for SIK1 and enhanced the time-dependent inhibition (TDI) properties of CYP by modulating the structural characteristics of CYP. In immune-mediated in vitro and in vivo models, GLPG3970's dual mechanism of modulating pro-inflammatory and anti-inflammatory cytokines has been confirmed.
GLPG3970 is a dual SIK2/SIK3 inhibitor that has been evaluated in clinical trials for autoimmune and inflammatory diseases. The authors improved the selectivity for SIK1 and enhanced the time-dependent inhibition (TDI) properties of CYP by modulating the structural characteristics of CYP. In immune-mediated in vitro and in vivo models, GLPG3970's dual mechanism of modulating pro-inflammatory and anti-inflammatory cytokines has been confirmed. Sequence alignment of the three SIK subtypes indicates that there are only five non-conserved residues in the ATP-binding site, suggesting a high degree of similarity between subtypes. Visual inspection of the ligand-binding sites in both sequence alignment and X-ray structures was used to predict the impact of different residues on SIK selectivity. The remaining residues in the binding sites are conserved. SAR optimization identified a second methoxy group on the alkyl pyrazole and benzene ring as an important feature for high SIK potency. Residues surrounding the dimethoxybenzamide portion of compound 8 are very similar among isomers. In contrast, differing residues between SIK subtypes are located around the pyrazole substituent at the 5-position of the benzimidazole scaffold in compound 8. Notably, tyrosine residues in SIK2 (Tyr98) and SIK3 (Tyr144) are replaced by a phenylalanine residue in SIK1 (Phe105). The pyrazole moiety in compound 8 is coplanar with the benzimidazole scaffold, allowing for displaced π-π interactions with either tyrosine or phenylalanine, and forming a weak hydrogen bond between the mildly polarized pyrazole C-H group and the carbonyl portion of alanine within the hinge region, resulting in pan-SIK inhibition. The authors conducted a preliminary exploration of modifications to the substituents and scaffolds. Replacement of the pyrazole ethyl group at the 5-position of the benzimidazole scaffold in compound 9 with a non-aromatic morpholine substituent (10) led to at least a 25-fold decrease in SIK activity, while compound 10 showed a trend toward selectivity for SIK1.
Sequence alignment of the three SIK subtypes indicates that there are only five non-conserved residues in the ATP-binding site, suggesting a high degree of similarity between subtypes. Visual inspection of the ligand-binding sites in both sequence alignment and X-ray structures was used to predict the impact of different residues on SIK selectivity. The remaining residues in the binding sites are conserved. SAR optimization identified a second methoxy group on the alkyl pyrazole and benzene ring as an important feature for high SIK potency. Residues surrounding the dimethoxybenzamide portion of compound 8 are very similar among isomers. In contrast, differing residues between SIK subtypes are located around the pyrazole substituent at the 5-position of the benzimidazole scaffold in compound 8. Notably, tyrosine residues in SIK2 (Tyr98) and SIK3 (Tyr144) are replaced by a phenylalanine residue in SIK1 (Phe105). The pyrazole moiety in compound 8 is coplanar with the benzimidazole scaffold, allowing for displaced π-π interactions with either tyrosine or phenylalanine, and forming a weak hydrogen bond between the mildly polarized pyrazole C-H group and the carbonyl portion of alanine within the hinge region, resulting in pan-SIK inhibition. The authors conducted a preliminary exploration of modifications to the substituents and scaffolds. Replacement of the pyrazole ethyl group at the 5-position of the benzimidazole scaffold in compound 9 with a non-aromatic morpholine substituent (10) led to at least a 25-fold decrease in SIK activity, while compound 10 showed a trend toward selectivity for SIK1.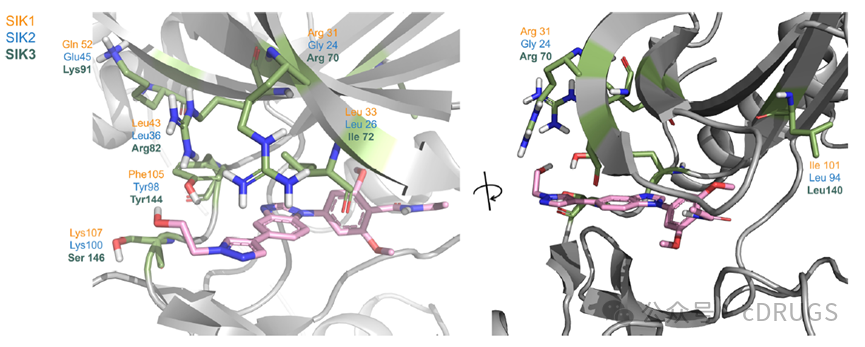
X-ray structure of SIK3 in complex with 8
Starting from compound 10, the authors explored scaffold hopping, morpholine replacements, and modifications of the benzamide moiety to enhance potency against SIK and selectivity for SIK1. The scaffold hop from benzimidazole in 10 to imidazo[1,2-a]pyridine in 11 increased potency against SIK2 and SIK3 while improving selectivity for SIK1. Replacing one methyl group of the dimethoxyphenyl moiety with difluoromethyl and the trifluoroethyl of the carboxamide portion with cyclopropyl led to compound 12. These changes further enhanced the potency against SIK2 and SIK1. Using molecules 11 and 12 as a basis, paired tests were conducted on the imidazo[1,2-a]pyridine scaffold with other amine substituents. The tested substituents were difluoroazetidine (13 and 14) and N-methylpiperazine (15 and 16), which showed improved potency, particularly against SIK2. Compared to 13, compound 14 exhibited stronger inhibitory activity against SIK1, SIK2, and SIK3, as observed with N-methylpiperazine derivatives 16 and 15. Although 16 demonstrated robust activity against SIK2 and SIK3 and exciting selectivity for SIK1, its progression was halted in the CYP TDI assay using human liver microsomes due to changes in IC50. This characteristic is undesirable as it may lead to drug-drug interactions (DDI) and off-target drug toxicity.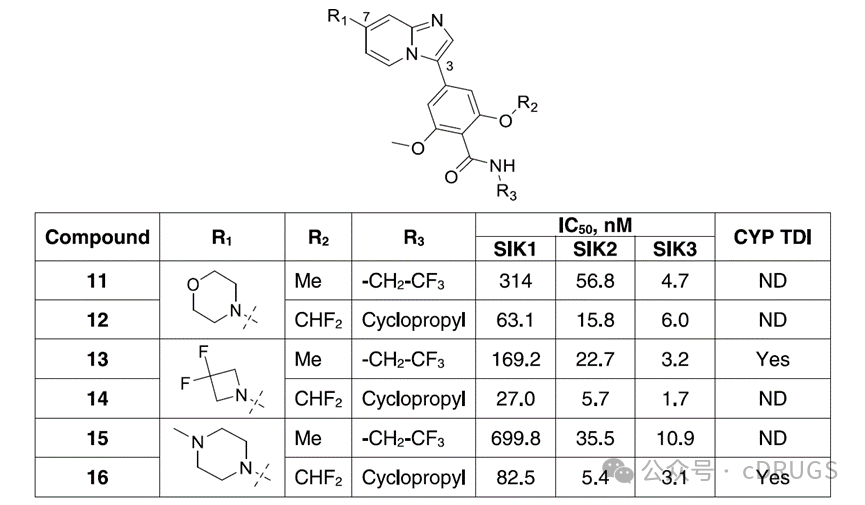 To enhance the selectivity for SIK1, the authors further explored the amine substitution at the 7-position of the imidazo[1,2-a]pyridine scaffold using cyclopropylamine. Replacing the N-methyl basic center of compound 16 with the polar non-basic acetamide group in compound 17 maintained similar potency on SIK, and extending the chain on the piperazine nitrogen of compound 18 to a hydroxyethyl group also retained equivalent potency. Introducing a basic dimethylamino group at the 4-position of the piperidine ring substituent (compound 19) preserved the potency for SIK2 and SIK3 but reduced selectivity for SIK1. In contrast, analog 20, bearing a dimethylamino group on an azetidine substituent, retained potency and selectivity comparable to the methylpiperazine derivative 16. These data suggest that the position of the basic group plays a crucial role in SIK1 selectivity. However, compound 20 tested positive in the CYP TDI assay and was not pursued further. Replacement of the imidazo[1,2-a]pyridine scaffold of compound 21 with an aniline moiety or a heteroaromatic amine as in compound 22 resulted in activity on SIK without subtype selectivity. Compounds 23 and 24, containing an amide substituent, maintained effective activity on SIK3 with selectivity for SIK1 but showed reduced activity on SIK2. The incorporation of a flexible acyclic linker between the imidazo[1,2-a]pyridine scaffold and the basic group was tolerated, as exemplified by compound 25, which contains a 2-(morpholino)ethanamine substituent and displays low nanomolar potency for SIK2 and SIK3 with some selectivity for SIK1. Replacing the nitrogen in the linker with oxygen as in the ether analog 26 resulted in single-digit nanomolar activity for SIK2 and SIK3 and 10-fold and 30-fold selectivity for SIK1, respectively. Similarly, replacing the morpholine moiety with a tetrahydropyran derivative 27 led to a reduction in SIK1 selectivity to 4-fold/10-fold. An important observation from the evaluation of the CYP TDI assay was that while the N-linked derivative 25 tested positive in the CYP TDI assay, the O-linked analog 26 tested negative, and the tetrahydropyran analog 27 tested positive. The authors speculated that this might be related to the imidazo[1,2-a]pyridine scaffold, as it is known to possess structural alerts associated with the risk of forming reactive metabolites and causing CYP TDI. Strategies to eliminate CYP TDI include modifying its physicochemical properties, altering the stereoelectronic properties of substituents, and introducing soft spots to redirect metabolism to another site. Data suggest that the increased lipophilicity of compound 27 (CLogP = 4.4) compared to compound 26 (CLogP = 3.8) restored CYP TDI activity. Since the ether linker can improve DDI performance, it was retained for further study, and the lipophilicity of the compounds was monitored.
To enhance the selectivity for SIK1, the authors further explored the amine substitution at the 7-position of the imidazo[1,2-a]pyridine scaffold using cyclopropylamine. Replacing the N-methyl basic center of compound 16 with the polar non-basic acetamide group in compound 17 maintained similar potency on SIK, and extending the chain on the piperazine nitrogen of compound 18 to a hydroxyethyl group also retained equivalent potency. Introducing a basic dimethylamino group at the 4-position of the piperidine ring substituent (compound 19) preserved the potency for SIK2 and SIK3 but reduced selectivity for SIK1. In contrast, analog 20, bearing a dimethylamino group on an azetidine substituent, retained potency and selectivity comparable to the methylpiperazine derivative 16. These data suggest that the position of the basic group plays a crucial role in SIK1 selectivity. However, compound 20 tested positive in the CYP TDI assay and was not pursued further. Replacement of the imidazo[1,2-a]pyridine scaffold of compound 21 with an aniline moiety or a heteroaromatic amine as in compound 22 resulted in activity on SIK without subtype selectivity. Compounds 23 and 24, containing an amide substituent, maintained effective activity on SIK3 with selectivity for SIK1 but showed reduced activity on SIK2. The incorporation of a flexible acyclic linker between the imidazo[1,2-a]pyridine scaffold and the basic group was tolerated, as exemplified by compound 25, which contains a 2-(morpholino)ethanamine substituent and displays low nanomolar potency for SIK2 and SIK3 with some selectivity for SIK1. Replacing the nitrogen in the linker with oxygen as in the ether analog 26 resulted in single-digit nanomolar activity for SIK2 and SIK3 and 10-fold and 30-fold selectivity for SIK1, respectively. Similarly, replacing the morpholine moiety with a tetrahydropyran derivative 27 led to a reduction in SIK1 selectivity to 4-fold/10-fold. An important observation from the evaluation of the CYP TDI assay was that while the N-linked derivative 25 tested positive in the CYP TDI assay, the O-linked analog 26 tested negative, and the tetrahydropyran analog 27 tested positive. The authors speculated that this might be related to the imidazo[1,2-a]pyridine scaffold, as it is known to possess structural alerts associated with the risk of forming reactive metabolites and causing CYP TDI. Strategies to eliminate CYP TDI include modifying its physicochemical properties, altering the stereoelectronic properties of substituents, and introducing soft spots to redirect metabolism to another site. Data suggest that the increased lipophilicity of compound 27 (CLogP = 4.4) compared to compound 26 (CLogP = 3.8) restored CYP TDI activity. Since the ether linker can improve DDI performance, it was retained for further study, and the lipophilicity of the compounds was monitored.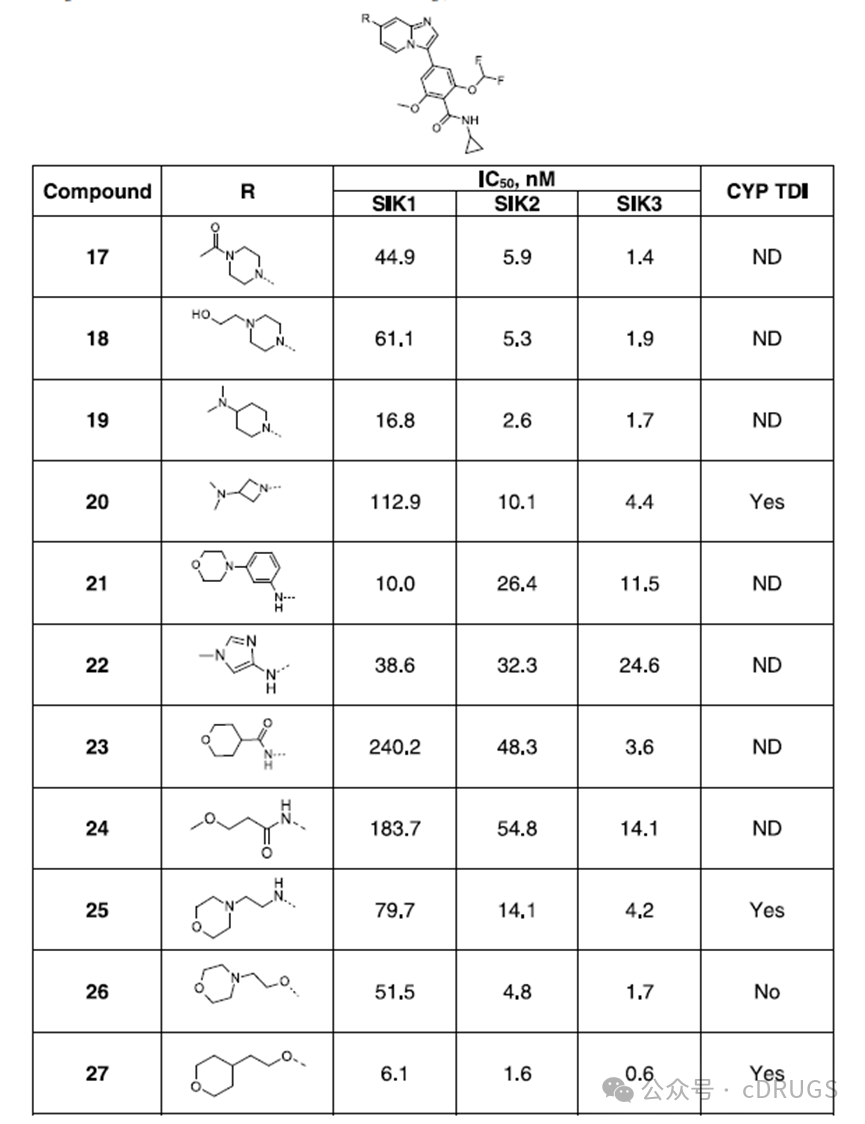 Although the residues around the dimethoxybenzamide moiety at the active site are highly conserved among SIK subtypes, it cannot be ruled out that more distant non-conserved residues may lead to subtle conformational differences in the active sites between subtypes, thereby promoting subtype selectivity. Therefore, the authors investigated the impact of modifications to the benzamide portion of the ether derivative 26 on the potency against SIK and its selectivity for SIK1. The phenyl ring was replaced with a pyridine ring because the pyridine ring can form intramolecular hydrogen bonds, which is detrimental to potency. Compounds 28 and 29, containing cyclopropyl amide, and compound 30, containing trifluoroethyl amide, exhibited IC50 values above 50 nM on all three SIK isoforms and showed some selectivity for SIK1. Compared to the pyridine analog 28, cyclization of the amide with one of the meta positions yielded derivative 31, featuring a cyclopropyl-substituted lactam ring, thereby enhancing potency and selectivity. Finally, introducing a trifluoroethyl group onto the lactam ring instead of the cyclopropyl group resulted in GLPG3970 (32), which demonstrated single-digit nanomolar potency on SIK2 and SIK3, with IC50 values of 7.8 nM and 3.8 nM, respectively, and selectivity for SIK1 of 36-fold and 74-fold (IC50 = 282.8 nM). Compound 32 displayed lipophilicity similar to compound 26 and tested negative in the CYP TDI assay.
Although the residues around the dimethoxybenzamide moiety at the active site are highly conserved among SIK subtypes, it cannot be ruled out that more distant non-conserved residues may lead to subtle conformational differences in the active sites between subtypes, thereby promoting subtype selectivity. Therefore, the authors investigated the impact of modifications to the benzamide portion of the ether derivative 26 on the potency against SIK and its selectivity for SIK1. The phenyl ring was replaced with a pyridine ring because the pyridine ring can form intramolecular hydrogen bonds, which is detrimental to potency. Compounds 28 and 29, containing cyclopropyl amide, and compound 30, containing trifluoroethyl amide, exhibited IC50 values above 50 nM on all three SIK isoforms and showed some selectivity for SIK1. Compared to the pyridine analog 28, cyclization of the amide with one of the meta positions yielded derivative 31, featuring a cyclopropyl-substituted lactam ring, thereby enhancing potency and selectivity. Finally, introducing a trifluoroethyl group onto the lactam ring instead of the cyclopropyl group resulted in GLPG3970 (32), which demonstrated single-digit nanomolar potency on SIK2 and SIK3, with IC50 values of 7.8 nM and 3.8 nM, respectively, and selectivity for SIK1 of 36-fold and 74-fold (IC50 = 282.8 nM). Compound 32 displayed lipophilicity similar to compound 26 and tested negative in the CYP TDI assay.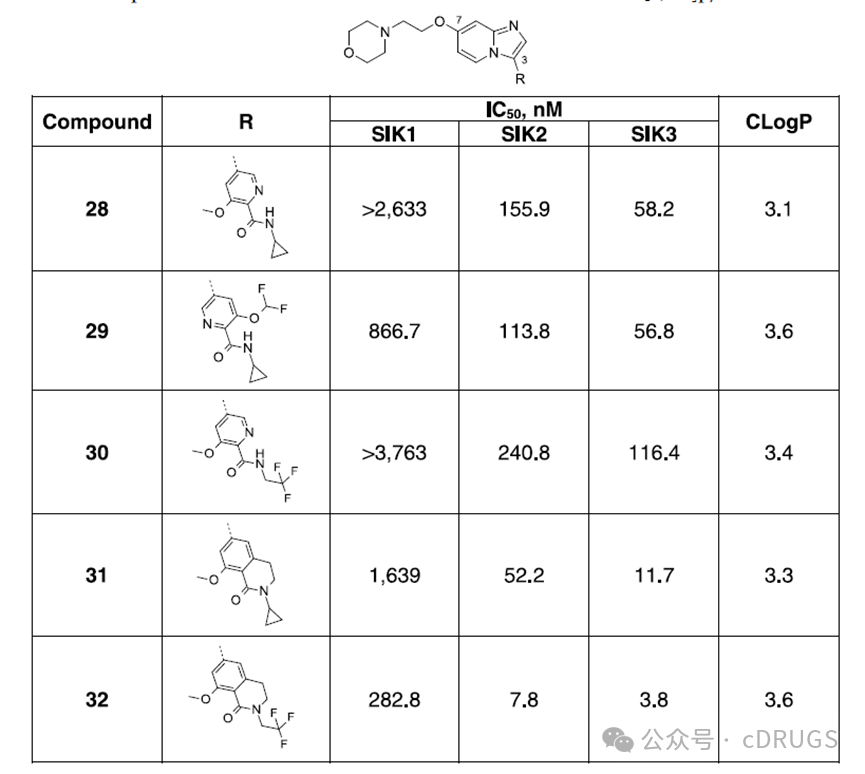 In summary, considering the co-crystal structure of compound 8 in SIK3 and the non-conserved residues surrounding the pyrazole group bound to the benzimidazole scaffold in different SIK subtypes, the authors were motivated to explore structural modifications in this region to achieve selectivity for SIK1. Starting from compound 10, which features a morpholine substituent on the benzimidazole scaffold instead of a pyrazole group, the compound exhibited moderate activity against SIK. Shifting from benzimidazoles to imidazo[1,2-a]pyridines and modifying the benzamide moiety enhanced the potency against SIK. Replacing the morpholine substituent with either N-cyclic or acyclic substituents containing basic groups effectively conferred activity against SIK2 and SIK3 while improving selectivity for SIK1. The oxygen chain substitution in analog 26 improved the TDI properties of CYP and showed effective activity against SIK2 and SIK3, though selectivity for SIK1 still needed enhancement. Finally, cyclizing the benzamide moiety into a trifluoroethyl-alkylated lactam yielded ether analog 32 (GLPG3970), which demonstrated the expected level of potency against SIKs and selectivity for SIK1, and tested negative in the CYP TDI assay.
In summary, considering the co-crystal structure of compound 8 in SIK3 and the non-conserved residues surrounding the pyrazole group bound to the benzimidazole scaffold in different SIK subtypes, the authors were motivated to explore structural modifications in this region to achieve selectivity for SIK1. Starting from compound 10, which features a morpholine substituent on the benzimidazole scaffold instead of a pyrazole group, the compound exhibited moderate activity against SIK. Shifting from benzimidazoles to imidazo[1,2-a]pyridines and modifying the benzamide moiety enhanced the potency against SIK. Replacing the morpholine substituent with either N-cyclic or acyclic substituents containing basic groups effectively conferred activity against SIK2 and SIK3 while improving selectivity for SIK1. The oxygen chain substitution in analog 26 improved the TDI properties of CYP and showed effective activity against SIK2 and SIK3, though selectivity for SIK1 still needed enhancement. Finally, cyclizing the benzamide moiety into a trifluoroethyl-alkylated lactam yielded ether analog 32 (GLPG3970), which demonstrated the expected level of potency against SIKs and selectivity for SIK1, and tested negative in the CYP TDI assay.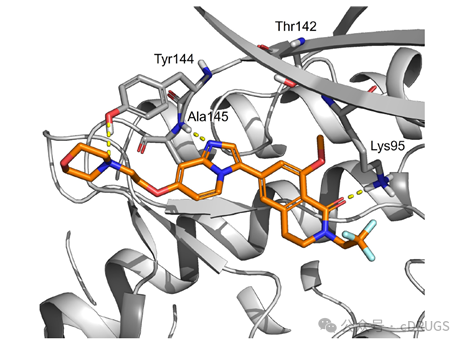
Representative frame from molecular
dynamics simulations of compound 32 in SIK3
To understand the selectivity of compound 32, docking and molecular dynamics simulations were performed in SIK3. Compound 32 binds to the protein at the ATP site, adopting an active conformation similar to that of compound 8, and can thus be classified as a Type I kinase inhibitor. In the simulation, the morpholine ring of compound 32 occupied the solvent-exposed region while maintaining hydrogen bonding interactions with Tyr144. This tyrosine residue is conserved in SIK2 but not in SIK1, suggesting that interaction with this residue may contribute to dual selectivity for SIK2/SIK3. Compound 32 demonstrated the expected efficacy in diseased mouse models and exhibited suitable pharmacokinetics, ADMET, and in vitro safety for further development.Original link: https://doi.org/10.1021/acs.jmedchem.3c02246
Long press to follow this official account
Reprint/Authorization/Academic CooperationPlease contact the Official Account Assistant.


