Eli Lilly Discloses First-in-Class Small Molecule Inhibitor of Lipoprotein(a) Assembly with Sub-nanomolar Potency

Eli Lilly
Global Pharmaceutical R&D and Production Company
On May 8, Eli Lilly published in Nature its targetedLipoproteinA Novel and Potent Inhibitor of ALY-3473329Molecular design.LY-3473329 is aWith sub-nanomolar potency (IC50=0.09nM) compounds that can inhibitLp(a)The formation. This article is on pure pharmaceutical molecular design and optimization published inNatureThe main journal, many of its strategies are still worth learning, especially for the design of protein interaction inhibitors.
Eli Lilly's this productLp(a) InhibitorLY-3473329 As of2024-04-20(NCT06342596) CompletedIIPhase ClinicalRecruitment(Global233People, including mainland China), used to treat hyperlipoproteinemia(a)Hemopathy。

1. Background Introduction
Lp(a) It is an independent cardiovascular risk factor determined by genes, which cannot be changed by altering lifestyle, and there are no medications that can specifically reduce it.Lp(a) Horizontally without causing other effects. Approximately20% High serum levels exist in the population.Lp(a) Level, leading to an increased risk of the first cardiovascular event beyond1.6 Times, the second cardiovascular event exceeds1.42 Times, the risk increase is associated with serumLp(a) Proportional to the level.Lp(a) The circulating level is mainly determined byKringle IV Subtype2 (KIV 2) Domain andLPA GeneDecision. Epidemiological studies have identifiedLp(a)Is an independent cardiovascular risk factor, and due toLp(a)Levels are determined by genetic structure.Lp(a)Diseases that have the effect of causing atherosclerosis and aortic valve calcification.
Lp(a) It is a lipoprotein particle formed through a two-step process, involvingLDL Particles and another proteinapo(a) Interactions between.The1Step,apo(a) Through a uniqueK IV 5-8(Structural elements of the lysine binding site), andLDL On the particlesapoB-100 The region rich in lysine non-covalently binds. Among them, withapoB The lysine coordination residue is homologous.The2Step,apo(a) TheK IV 9 UnpairedCys67 Residue andapoB-100 TheCys3734BetweenFormation of Disulfide Bonds,Covalently linking two proteins。
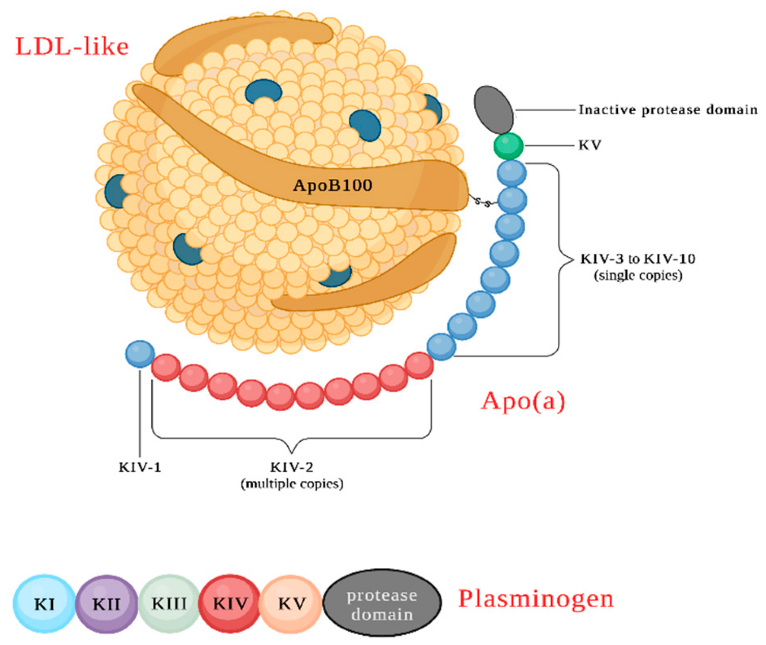
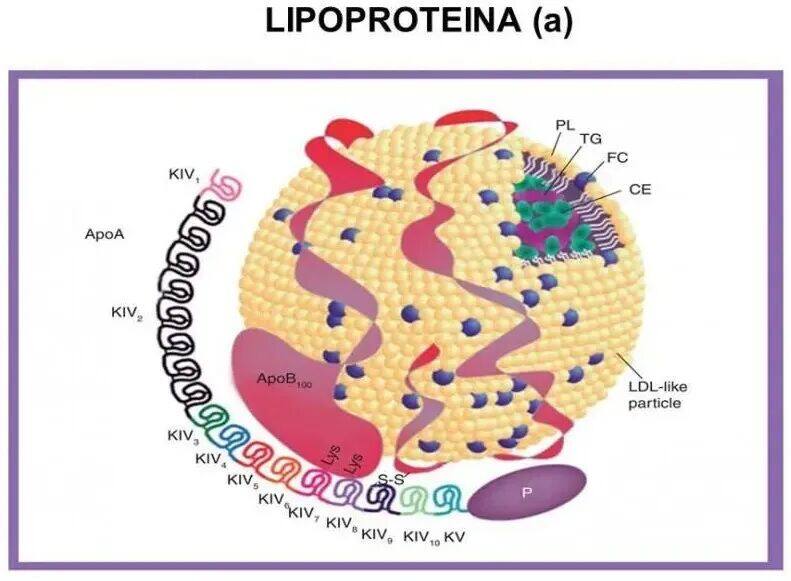
Lp(a) Structure and Composition. Lp(a) Added byApo(a) The composition of low-density lipoprotein inApo B100 AndApo(a) Form disulfide bonds betweenKey。 Apo(a) The protein structure is composed of many functional domains. Overall, its structure is very similar to that of plasminogen, a zymogen whose conversion to plasmin determines the activation of the thrombolysis process. Plasminogen also exhibits a series ofCyclic Structure (K,kringle) `, numbered as `I ToV, followed by a serine protease-like catalytic domain, which can be cleaved by tissue plasminogen activator and urokinase to produce plasmin.Inapo(a)In China,Some domains are very similar to the analogous domains of plasminogen, including the carboxyl-terminal catalytic domain.The proteolytic domain, but it cannot be activated like plasminogen. apo(a)The uniqueness lies inkringle IV The number of repetitions is relatively high; however, these repetitions do not produce the same structure, but rather producekringle IV Subtype, from type1 To Type10(type)。
Apo(a) Found only in the genomes of humans and some non-human primates.LPA Gene by plasminogen(PLG) Genes are produced through gene duplication in the later stages of evolution, where plasminogenKringle DomainI、II AndIII Lost, andKIV The domain extends and differentiates into10Subtype(K IV 1–10)(SupplementFigure 1); therefore,apo(a) TheK IV Domain and PlasminogenKringle The domain has70-85% Sequence identity.
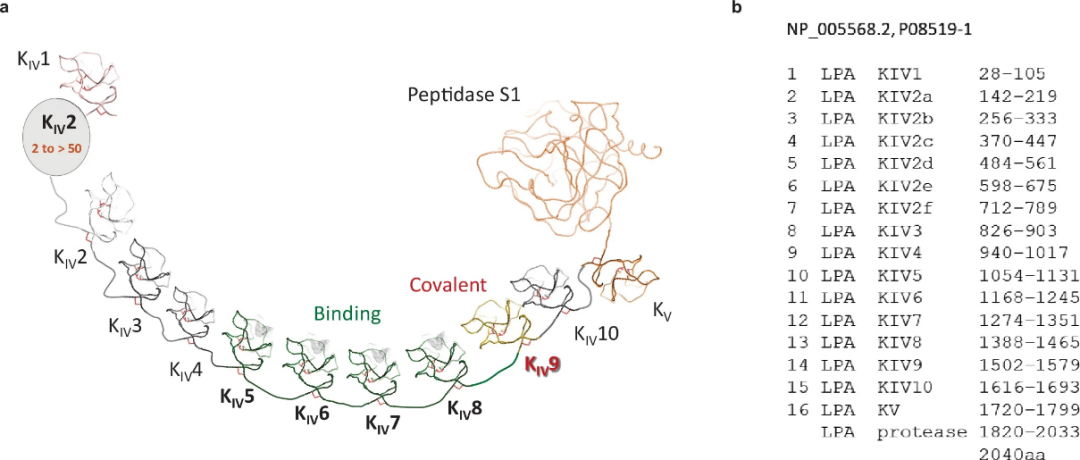
SupplementFigure 1. apo(a) TheK IV Domain
Currently, there is no specific method to reduce Lp(a) Horizontal treatment methods.Pelacarsen(Targeting LPA The antisense oligonucleotide) andolpasiran(A kind of LPA Small InterferenceRNA) are currently in3 Injectable therapy in Phase III clinical studies.
HereDevelopment of an effective, selective oral small-molecule therapeutic agent by blocking apo(a) AndapoB The first interaction between to inhibitLp(a) Formation, thereby reducingLp(a) Circulating Levels。Strategies Focus on Identifying Binding apo(a) KIV 7-8 molecules to inhibitapo(a) AndapoB The initial interaction between. Mainly becauseReorganization apo(a) In vitro studies of mutant proteins indicateapo(a) ThroughK IV 6-8WithapoB-100 The lysine-rich region is non-covalently bound.
2. Emerging Molecule Discovery
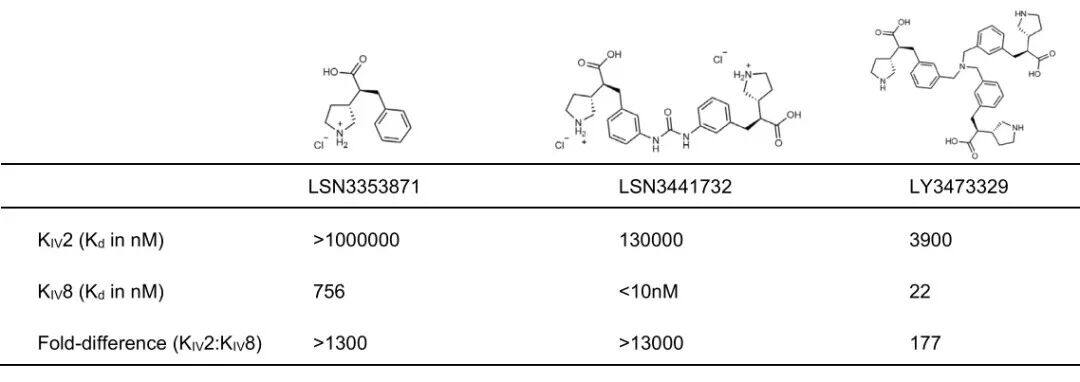
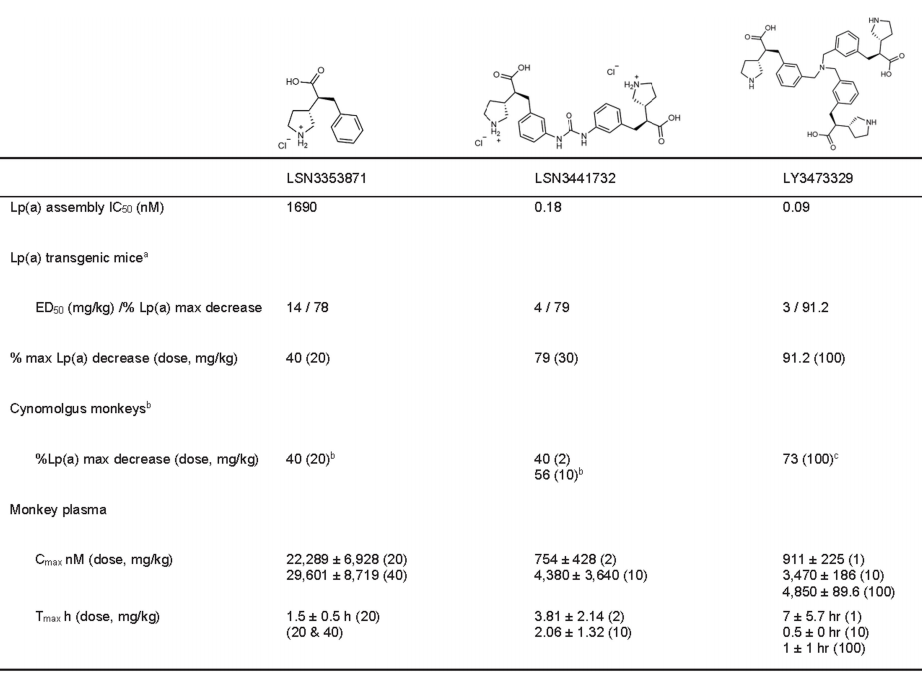
Using purified recombinant apo(a) KIV 7-8 Protein ProgressBiochemical and Biophysical Compound Screening(The specific plan has not been disclosed. It has a simple structure. The editor speculates that it might be similar to DEL or affinity-based mass spectrometry screening.), identifying interacting small molecules. Optimization of the initial binding molecule led to LSN3353871 The development and discovery, which combinesK IV 8、K IV 7–8 AndK IV 5–8。Through isothermal calorimetry(ITC) Measurement,K d The values are respectively756 nM、605 nM And423 nM(Figure 1a、b). In contrast,LSN3353871 Not bindingK IV 2(Table 1). In vitroLp(a) In the assembly measurement,LSN3353871 DestroyedLp(a) Formation,IC 50For 1.69 µM(Figure 1c) . Crystal structure analysis showsLSN3353871AndKIV 8Amino acid residueGlu56、Asp54、Tyr62AndArg69Interactions illustrate the specificity and binding mode of the molecule (Figure1d). For expressing individuals Lp(a) Oral administration in miceLSN3353871,Lead toLp(a) Levels decreased in a dose-dependent manner by up to78%`, Median Effective Dose`(ED50 ) 14 mg kg−1 Twice Daily(BID). In cynomolgus monkeys, administration ofLSN3353871(20 mg·kg−1 BID, lasting for two weeks) led toLp(a) Decrease by up to40%(Figure 1e,f). The difference in the degree of influence between these two models can be explained by Lp(a) Differences in expression levels and metabolism may explain this, as the mouse genome itself does not containLPA Genes. These data provide a proof of concept thatapo(a) Small molecules binding to the lysine-binding domain can inhibit in vivoLp(a) Formation.
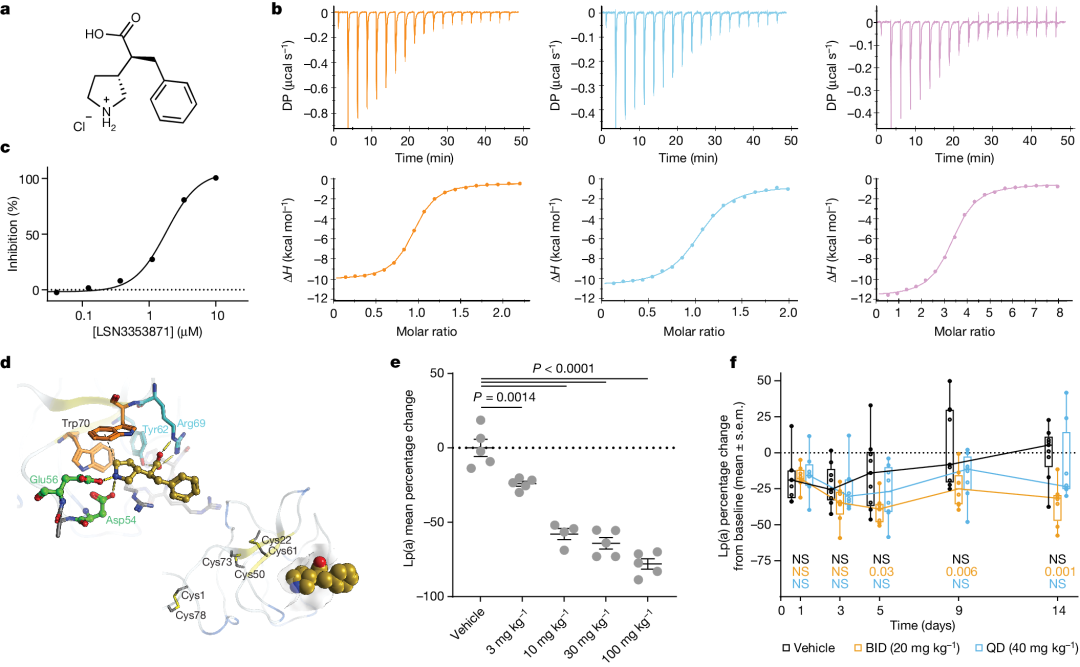
Figure 1: Small moleculeapo(a) Binder andLp(a) Identification of Forming Inhibitors.
2. Lead Compound Discovery
WillIncludeLSN3353871And K IV Domain InteractionStructureOfMutualConnectionGeneratedBivalent MoleculeLSN3441732(Figure2a). This dimeric compound inhibits in vitro Lp(a) Formation of particles,IC50 =0.18 nM, with the maternal monomer moleculeLSN3353871Compared to the efficacy, it has increased by three orders of magnitude.(IC50 = 1.69µM), Compared with the optimized monomer ligand LSN3374443 (IC 50 = 36 nM) Increased by two orders of magnitude in comparison.(Figure2b、c)。This is quite interesting. Does it also affect the dimerization of the protein?
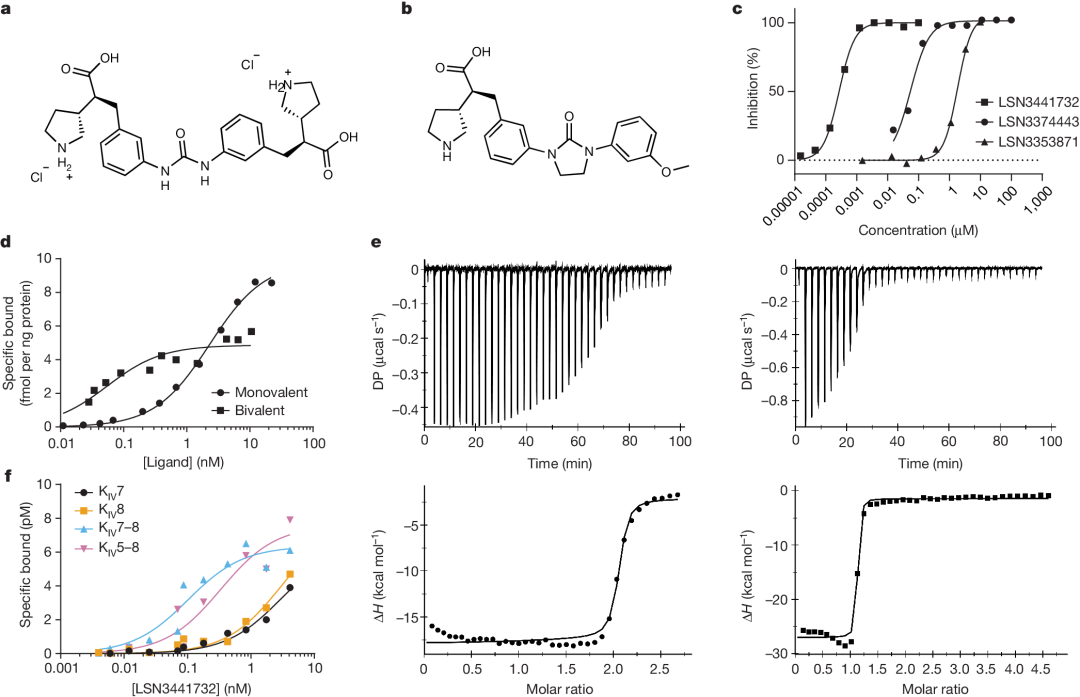
Figure 2: Withapo(a) K IV Mechanism of Domain Interaction and MultivalencyMoleculeDiscovery.
In order to determine multivalent and monovalent ligands apo(a) KIV Combined InteractionDifference, using direct radioligand binding and ITC Method. For radioligand binding,SynthesisDimerization 3 H-LSN3441732 AndMonomer 3H-LSN3374443 Radioligand.
Except KIV 10 Externally, 3 H-LSN3374443 Compared with all the tests containingK IV The protein binding, with an affinity similar to that of the full-lengthapo(a) The combination is similar (K d =15–31 nM). In the direct binding assay, 3 H-LSN3374443 With full-length recombinant apo(a) Combination, AverageKd =2.6 nM,Maximum Saturable Binding (Bmax ) Value is9.8 fmol/ng's apo(a), equivalent toEach apo(a) Molecules have two to three ligands(Figure 2d). In contrast, 3 H-LSN3441732 Compared with the full length apo(a) The binding affinity has increased.50 Times,Kd = 0.05 nM, and with Bmax = 4.9 fmol/ng apo(a), indicating eachapo(a) The molecule has a ligand (Figure 2d). Use apo(a) K IV 5–8 Direct ligand binding experiments on the tandem domain showed the same potency and stoichiometric relationship; 3H-LSN3374443 Is less effective than 3H-LSN3441732,K d The values are respectively13 nM And0.33 nM. Compared with the full lengthapo(a) Consistent with the radioligand binding studies,apo(a) K IV 5-8 Tandem DomainITC Data showsLSN3374443 In2:1 Stoichiometric binding, andLSN3441732 In1:1 Stoichiometric Binding(Figure 2e); The combination of two compounds Kd <15 nM. MultivalentCompound3 H-LSN3441732 ToKIV 7–8 (Kd = 0.21 nM) AndKIV 8(K d = 0.33 nM), belowSinglePrice Compound KIV 7 (K d = 2.8 nM) AndKIV 8 (Kd = 2.0 nM), confirming that multivalent leads to a significant increase in efficacy (Figure 2f)。LSN3441732 The crystal structure analysis shows that multipleapo(a) K IV Domains can bind simultaneously, resulting in binding affinity andLp(a) Improved efficacy inhibition (SupplementFigure 2b). ITC experiments showedLSN3441732 Not ApplicableKIV 2 Combination (Table 1)。
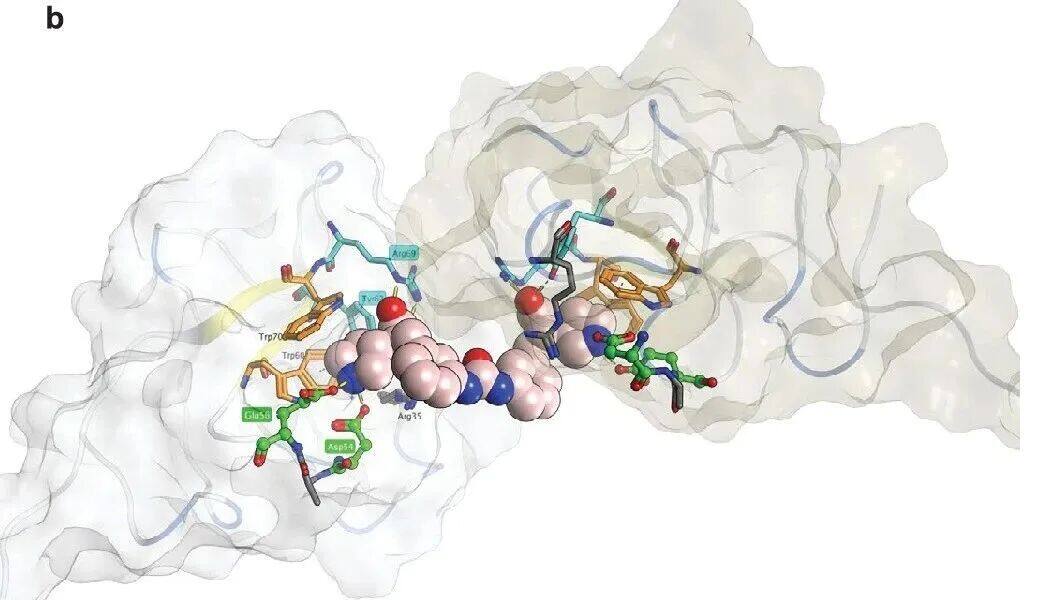
SupplementFigure2b. Crystal structure of bivalent inhibitors with proteins.
In Lp(a) In transgenic mice,LSN3441732 Plasma concentration increased in a dose-dependent manner after oral administration.BID Steady-state plasma after five days of dosingLp(a) Horizontal decline,ED50 For4 mg kg−1 ,30 mg kg −1 The average maximum inhibition rate at the dose79%(Figure 3a). In cynomolgus monkeys,2 mg kg −1 And10 mg kg −1 OralBID Dose ofLSN3441732 Later, MedianLp(a) The levels decreased by the largest margin respectively.45% And57%. Drug administration5 Days, and continuously inhibitLp(a) The steady-state level reached14 Day (Figure 3b)。 LSN3441732 Compatible with multivalent binding mechanisms, showing improved potency and efficacy both in vitro and in vivo compared to monovalent inhibitors.
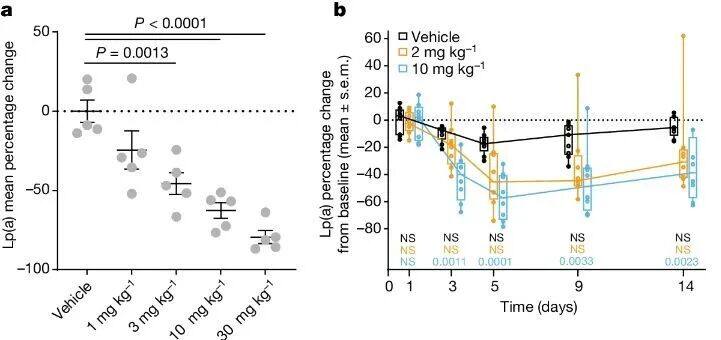
Figure 3: a-b, PK of bivalent inhibitors in vivo.
3. PCC Molecular Determination
To further leverage multivalency, synthesizedTrimer moleculeLY3473329(Figure3c)。 LY3473329 WithK IV 8 Analysis of the crystal structure of interactions between them indicates that trimeric molecules are able toCombined with three simultaneously K IV 8 Domain, which indicates more K IV apo(a) There may be domain interactions in (Figure3D, Supplementary Figure 2C)。 LY3473329-HCl salt Selective BindingK IV 8 , efficacy is22 nM`, and inhibit in vitro`Lp(a) Formation of particles,IC 50 =0.09 nM(Figure 3e,Table 2.). InLp(a)In transgenic mouse models,LY3473329-HClThe plasma levels of salt increase in a dose-dependent manner, and withLY3473329-HClTreatment ReducedLp(a)The level, absolutelyED 50 3 mg kg −1 ,BID OralMaximum after five days of administrationLp(a) Reduce92%(Figure 3f). In cynomolgus monkeys, LY3473329 In100 mg kg −1 Once Daily(QD) Dose-dependent reduction in median in the cohortLp(a) As high as71%(Figure3g)。Multivalence allows multiple apo(a) K IV The binding of the domain significantly improvedLp(a) Reduced in vitro and in vivo modelsLp(a) Efficacy and potency of forming inhibitors.
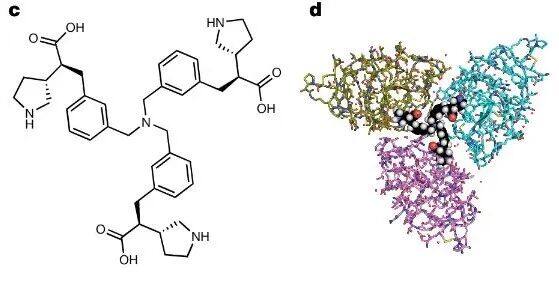
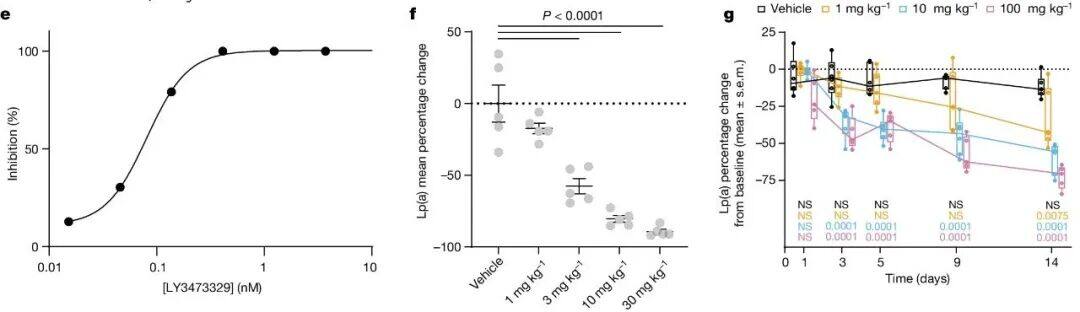
Figure 3: Trivalent InhibitorLY3473329Further increased forLp(a) Efficacy and effectiveness of forming inhibition.
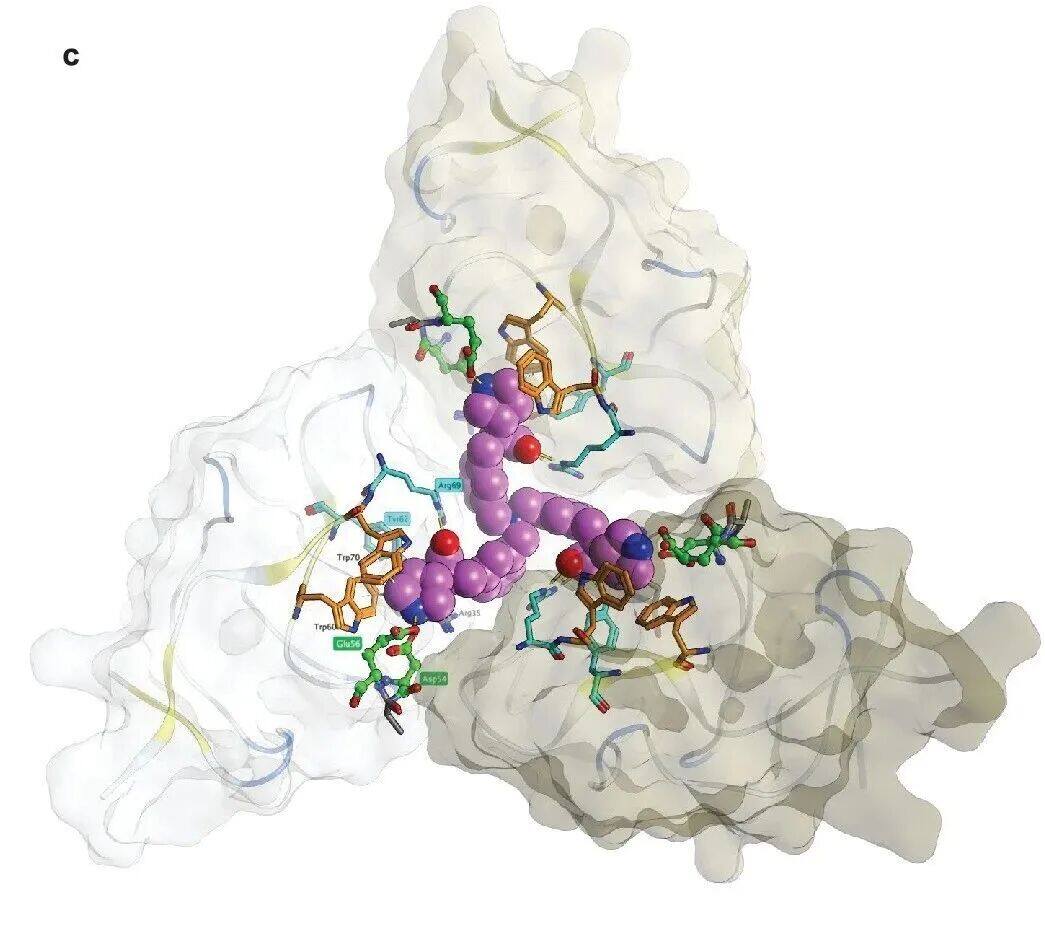
Supplementary Figure 2c. Crystal structure of the trivalent inhibitor.
4. PCC Molecular Safety Assessment - ToEffects of Plasminogen
Plasminogen is the zymogen precursor of plasmin, the primary fibrinolytic protease. Circulating plasminogen is comprised of a Pan-apple Domain, FiveKringle Domain(KI–V) Consists of a serine protease domain and adopts a closed, anti-activation conformation. The regulation of plasminogen activity is strictly controlled to ensure systemic hemostasis, and the inactivation of plasmin depends on its physiological inhibitors.α2-Antifibrinolytic enzymes, which remove free plasmin from circulation. Due toapo(a) KIV Domain and PlasminogenKringle The domain has a high degree of identity, thereforeNeedTo study whether these compounds can modulate plasminogen activity (measured via plasmin enzymatic activity).
Monomers and polyvalent compounds were added to rat and human plasma in vitro toEvaluate Possible Interference with Plasmin-Mediated Thrombolysis,But the activity of plasmin was not affected, which provided Lp(a) Evidence that inhibitors do not directly modulate plasminogen activation or plasmin activity.Despite the use of monomeric compoundsLSN3353871ProcessingRatsIn vivoNo inhibitory effect on plasmin activity was observed, butExtracorporealMeasurement,AndPolyvalent CompoundLSN3441732AndLY3473329-HClSalt reduces plasma plasmin activity (Figure4a,b). Since adding these compounds to plasma did not affect the enzymatic activity of plasmin in vitro, the mechanisms that might explain the reduced plasmin activity in animal models were investigated. In rats, daily oral administration of multivalent Lp(a) InterferentLSN3441732 Later, the liverPlg mRNA The level has not changed; however,LSN3441732AndLY3473329-HClSalt reduces plasma levels of plasminogen protein in a dose-dependent manner (Figure4a,b). Since these results raised the issue of multivalence Lp(a) InhibitorPossibility of reducing human plasmin activity, thus investigating the drivers of these compound-mediatedPotential Mechanisms of Decreased Plasminogen Levels。
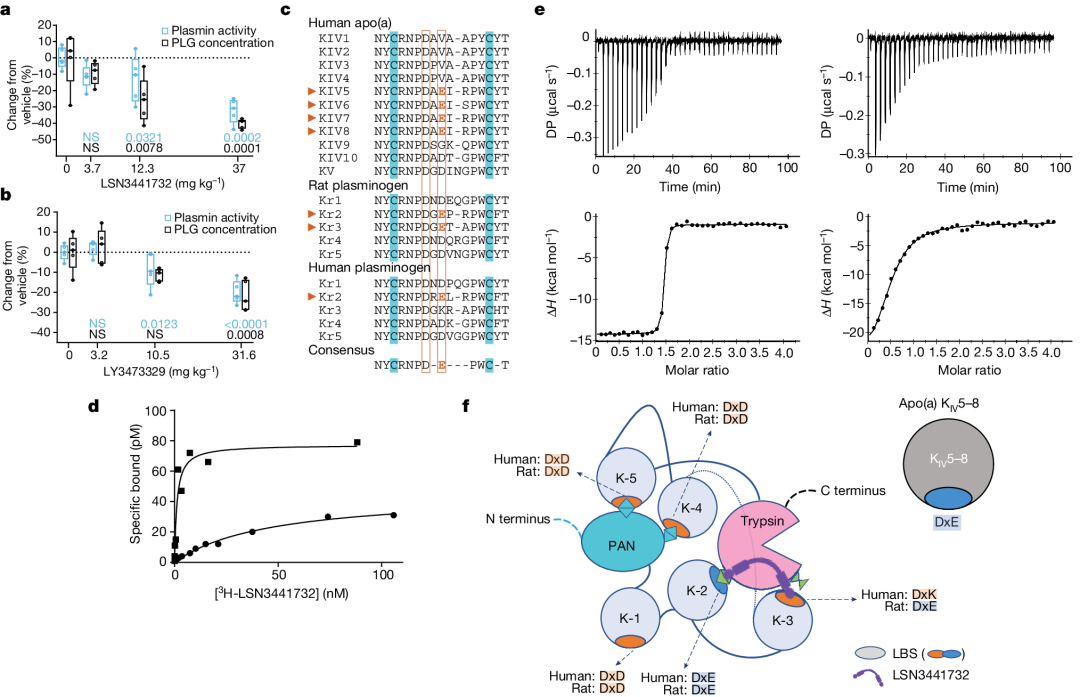
Figure 4: Selectivity for plasminogen.
Comparison of amino acid sequences of plasminogen in rats, cynomolgus monkeys, and humans (Figure 4c), and understand the closed anti-activation conformation Kringle The accessibility of the lysine binding site (as shown in the structure of human plasminogen),We assume Multivalent compounds exhibit species-specific plasminogen binding properties. LSN3353871The structural data revealedKIV8Aspartic acid (D), any amino acid (x) and glutamate residue (E)(DxEMolecular interactions between key motifs composed of motifs (Figure1d), Therefore, pay attention to the presence of these motifs in plasminogen. It is worth noting that, in apo(a) In China, fromKIV5 ToKIV8 Each ofKIV Repeatedly IncludedDxE Motif. RatPlasminogenKringle (K) K-2AndK-3Domains all containDxEMotif, and this motif exists only in human plasminogen.K-2In (Figure4c)。 Therefore,Assuming multivalent compounds can act as rat plasminogen in K-2 AndK-3 The bivalent binders, which can only bind throughK-2 The exclusive interaction as a monovalent binder of human plasminogen. If this is correct, the binding affinity of multivalent compounds to rat plasminogen should be higher than that to human plasminogen. Bivalent3H-LSN3441732Saturation binding experiments with molecules and rat and human plasminogen indicate,3H-LSN3441732Affinity for rat plasminogen compared to human plasminogenHigh50Times(Figure4d)。 According toITCEvaluation, the bivalent molecule with rat plasminogenK-2ToK-3The binding affinity of the domain is higher than that of human plasminogen.K-2ToK-3DomainHigh100TimesAbove; RatK-2ToK-3OfKdValue less than10nM, and peopleK-2ToK-3TheKdThe value is1.6μM(Figure4e)。 Indicates,Multivalent molecules and K-2 ToK-3 The result of plasminogen binding in rats disrupted the closure found in plasminogen.K-2–Lys708 Interact and trigger an open conformation that favors rapid degradation (Figure4f), thereby explaining the use ofLSN3441732 AndLY3473329-HCl Salt treatment reduces plasmin activity in animals.
In LY3473329 (muvalaplin) of the human body1 Evidence can be found in the study, indicating differences in the binding efficacy of plasminogen between rats and humans., leading to in the human bodyIn order to reduce Lp(a) Horizontally without affecting plasmin activity.
5. Summary of Molecular Design
In summary,This articleReported the use of apo(a) RepeatK IV Domain Structure,And throughComposite MultivalentTransform (small molecule multivalent, causing protein polymerization, inhibitionLp(a) Formation),Discovered and optimized Lp(a) The formation of effective and selective small molecule inhibitors. By examining such compounds withapo(a) Mechanism of binding between, developFinishedIn the human body apo(a) Rather than plasminogen, it has selective molecules. It is of significant reference value for designing inhibitors of protein-protein interactions, and its high activity in vivo and in vitro also makes it a very promising drug molecule for treating hyperlipidemia and lipoprotein(a) disorders.
Thank you for your likes, and friends who forward and share.
The official account is open for reprinting, and we also welcome everyone to make requests, leave messages, and engage in discussions.


~~~
Diaz, N., Perez, C., Escribano, A.M.et al. Discovery of potent small-molecule inhibitors of lipoprotein(a) formation. Nature (2024)