New Oncology Drug Approvals in March 2024: A Global Overview

CARsgen Therapeutics
Developer of CAR-T Cell Immunotherapy Drugs

Johnson & Johnson
Healthcare Product Manufacturers, Health Service Providers

Bristol-Myers Squibb
Biopharmaceutical and Nutritional Product R&D and Sales

BeOne
Developer of Molecular Targeted and Immune Anti-Tumor Drugs

CStone Pharmaceuticals
Innovative Oncology Immunotherapy and Precision Medicine Drug Developer

Takeda
Biopharmaceutical Manufacturer

AbbVie
Innovative Drug Developer

Astellas
Pharmaceutical R&D Manufacturer

IASO Biotechnology
Cancer Treatment New Drug Developer

According to publicly available data, in March 2024, multiple targeted and immunotherapy drugs were approved for marketing both in China and internationally, covering various cancer types such as non-small cell lung cancer (NSCLC), urothelial carcinoma (UC), and melanoma. Among them, Tolorametinib is the first MEK inhibitor in China and the world's first and only targeted drug approved for the treatment of advanced melanoma with NRAS mutations.
The detailed drugs approved for marketing in March and newly approved indications are shown below.
2024.03
01
New Drug Express (Summary Edition)
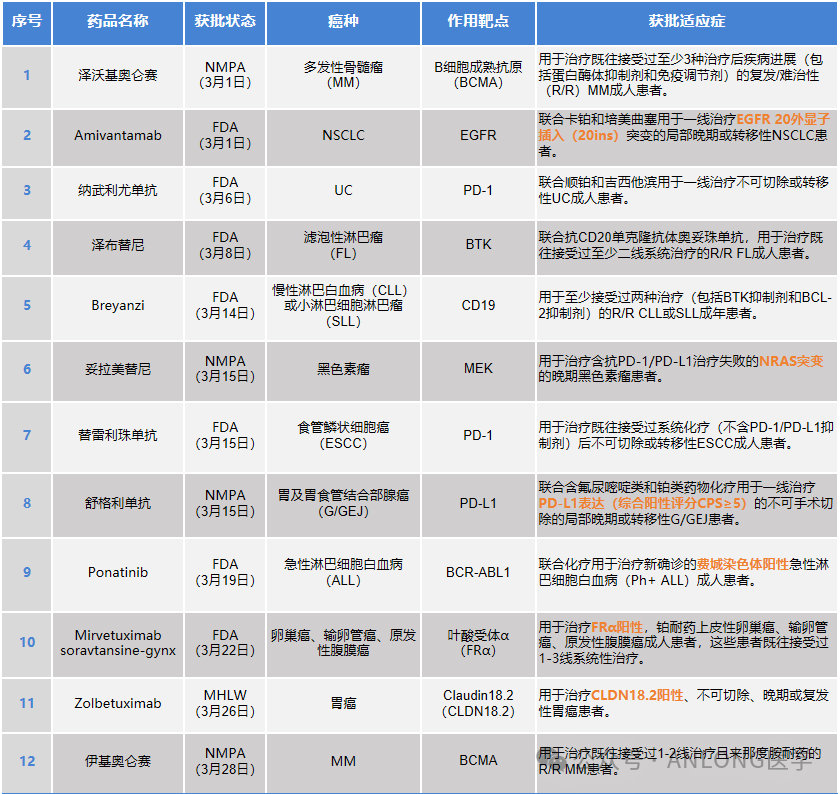
2024.03
02
New Drug Express (Detailed Version)
1. Zevor-cel
Product Name: Saikai Ze
Generic Name: Zevorcleucel
Indications:R/R MM
Clinical Trial:LUMMICAR STUDY 1
Original Research Company: CARsgen Therapeutics
Approval Date:2024.03.01
Approved Institution:NMPA
On March 1, 2024, CARsgen Therapeutics announced that the NMPA had approvedZevor-celMarketing Application,For the treatment of adult patients with R/R MM whose disease has progressed after receiving at least three prior therapies (including proteasome inhibitors and immunomodulatory agents).Zevokibart Celgene is the fifth CAR-T (Chimeric Antigen Receptor T-Cell) product to be launched in China and the second CAR-T product targeting BCMA.
This approval is based on an open-label, single-arm, multi-center, Phase II clinical study conducted in China.LUMMICAR STUDY 1(NCT03975907)In 2022, the American Society of Hematology (ASH) presented the data from this pivotal Phase II clinical trial. The study enrolled a total of 102 patients with R/R MM who had received ≥3 prior lines of therapy. As of August 16, 2022, all 102 patients had completed at least 3 months of follow-up or had withdrawn early, including 60 patients who completed at least 6 months of follow-up or withdrew early. The results showed that the median follow-up time for the 102 patients was 9 months.The objective response rate (ORR) was 92.2%, the rate of very good partial response (VGPR) and above was 85.3%, and the complete response/stringent complete response rate (CR/sCR) was 45.1%.Among the 60 patients treated, the median follow-up time was 12.1 months, with a CR/sCR rate of 56.7%. The median duration of response (DOR) and median progression-free survival (PFS) have not yet been reached. At the median follow-up of 9 months, the DOR rate was 86.1% and the PFS rate was 84.6%. In terms of safety, the injectable drug Zevor-cel was well-tolerated with controllable safety.
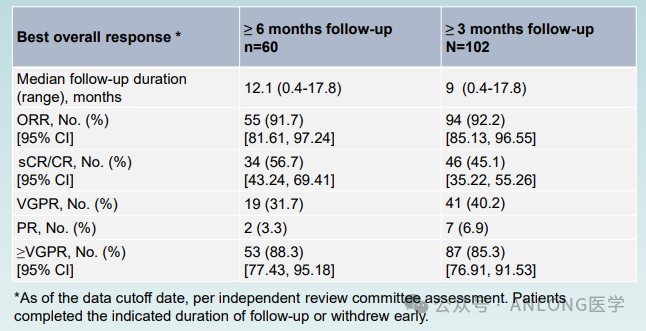
LUMMICAR STUDY 1 Data (from 2022 ASH)
MM is one of the most common hematologic system tumors, a malignant disease characterized by clonal proliferation of abnormal plasma cells. With the acceleration of aging in China and the increase in average life expectancy, the number of MM patients will continue to rise. According to estimates by Frost and Sullivan, the number of MM patients in China was approximately 153,000 in 2023, with 23,200 new cases. It is projected that the number of MM patients in China will grow to 266,300 by 2030. For newly diagnosed MM patients, commonly used first-line treatments include induction therapy, consolidation therapy, and maintenance therapy with various drug combinations, as well as autologous hematopoietic stem cell transplantation (ASCT), among others.
2. Amivantamab
Product Name:Rybrevant
Generic Name:Amivantamab
Indications:EGFR Exon 20 Insertion Mutation in NSCLC
Clinical Trial:PAPILLON
Original Research Company: Johnson & Johnson
Approval Date:2024.03.01
Approved Institution:FDA
On March 1, 2024, FDA approvedAmivantamabUnitedCarboplatin and PemetrexedUsed forFirst-line treatment for EGFR 20ins in patients with locally advanced or metastatic NSCLCPreviously, on May 21, 2021, Amivantamab was approved by the FDA for the treatment of locally advanced or metastatic NSCLC patients with EGFR 20ins mutations whose disease progressed during or after platinum-based chemotherapy. The indication has not yet been approved in China.
This approval is based on a randomized, open-label Phase III clinical study.PAPILLON(NCT04538664)The study aims to evaluate the efficacy and safety of Amivantamab combined with chemotherapy (carboplatin + pemetrexed) versus chemotherapy alone in newly diagnosed patients with advanced or metastatic NSCLC harboring EGFR 20ins. A total of 308 previously untreated patients with advanced or metastatic NSCLC carrying EGFR 20ins were enrolled and randomly assigned in a 1:1 ratio to either the Amivantamab plus chemotherapy group (n=153) or the chemotherapy-alone group (n=155). The primary endpoint of the study was progression-free survival (PFS) assessed by blinded independent central review (BICR); secondary endpoints included overall response rate (ORR), overall survival (OS), PFS after the first subsequent treatment (PFS2), and safety, among others. The results showed that, with a median follow-up of 14.9 months, the Amivantamab plus chemotherapy group and the chemotherapy-alone group had...The median PFS was 11.4 months vs. 6.7 months.,The PFS in the Amivantamab plus chemotherapy group was significantly prolonged by nearly 1 fold.The 18-month PFS rate was 31% vs. 3%, ORR was 73% vs. 47%, and median DOR was 9.7 months vs. 4.4 months., The median time to response (TTR) was 6.7 weeks vs. 11.4 weeks, with a mean tumor reduction of 53% vs. 34%. Safety analysis showed that the most common (≥40%) adverse events (AEs) in the Amivantamab plus chemotherapy group were neutropenia, paronychia, rash, anemia, infusion-related reactions, and hypoalbuminemia, with no new safety signals observed. The discontinuation rate of Amivantamab due to treatment-related adverse reactions was only 7%.
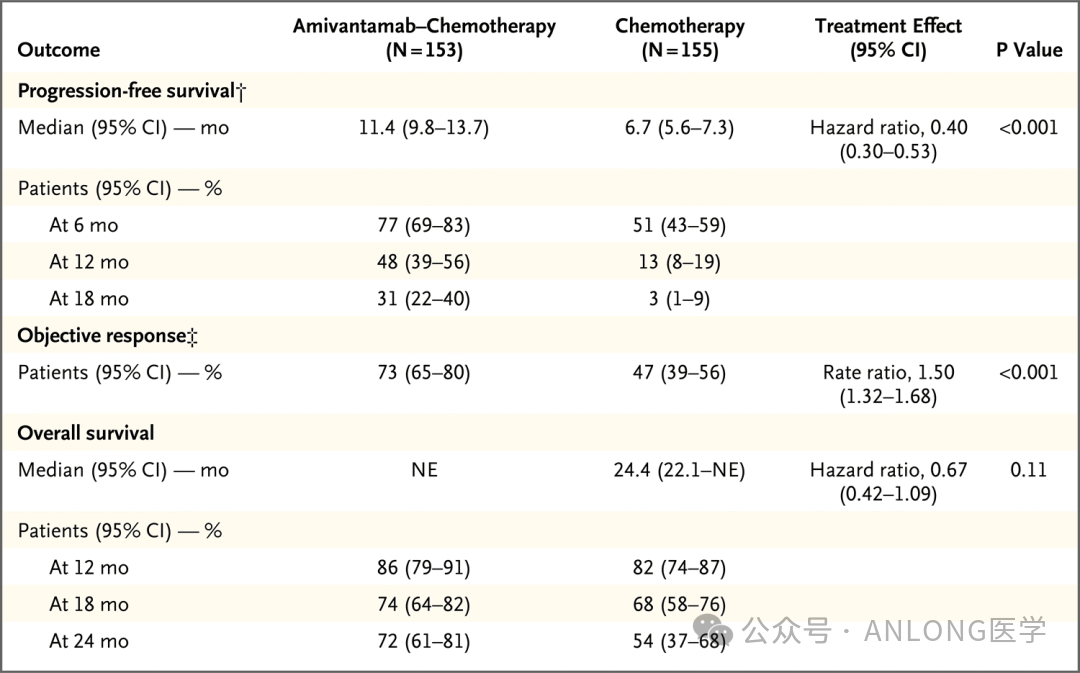
PAPILLON Study Data
3. Nivolumab
Product Name: Opdivo, Opdivo
Generic Name: Nivolumab, Nivolumab
Indications:UC
Clinical Trial:CHECKMATE-901
Original Research Company: Bristol-Myers Squibb
Approval Date:2024.03.06
Approved Institution:FDA
On March 6, 2024, FDA approvedNivolumabJointCisplatin and GemcitabineUsed forFirst-line treatment for adult patients with unresectable or metastatic UC.
This approval is based on a randomized, open-label Phase III clinical study.CHECKMATE-901(NCT03036098)The study enrolled a total of 608 previously untreated patients with unresectable or metastatic UC, who were randomly assigned in a 1:1 ratio to receive either nivolumab plus cisplatin and gemcitabine (for up to 6 cycles) followed by nivolumab monotherapy (for 2 years), or cisplatin plus gemcitabine (for up to 6 cycles). In both groups, patients discontinuing cisplatin could receive carboplatin. Randomization was stratified by tumor PD-L1 expression and liver metastasis. The primary efficacy endpoints were OS and PFS assessed by blinded independent central review according to RECIST v1.1. The results showed that the group receiving nivolumab combined with cisplatin and gemcitabine demonstrated statistically significant benefits in both OS and PFS compared to the group receiving cisplatin and gemcitabine alone.The median OS was 21.7 months for patients treated with nivolumab plus cisplatin and gemcitabine, compared to 18.9 months for those treated with cisplatin and gemcitabine alone. The median PFS was 7.9 months and 7.6 months, respectively.In terms of safety, the most common adverse reactions (≥15%) in patients receiving nivolumab combined with platinum-based doublet chemotherapy are nausea, fatigue, musculoskeletal pain, constipation, decreased appetite, rash, vomiting, etc.
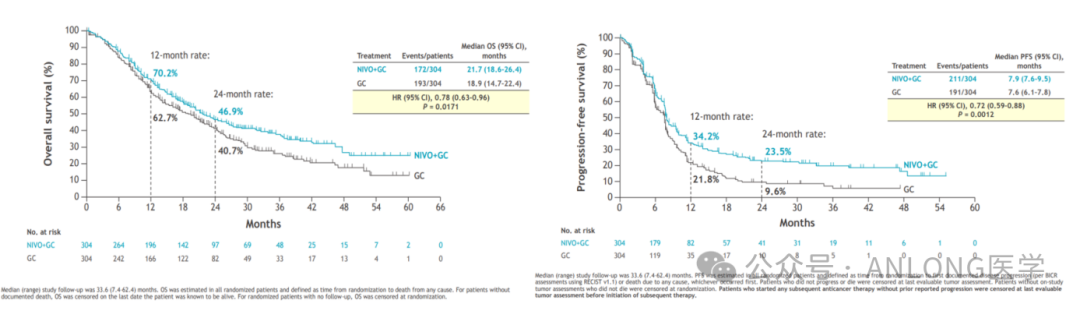
Median PFS and OS Results from the CHECKMATE-901 Study
4. Zanubrutinib
Product Name:Baiyueze, Brukinsa
Generic Name:Zanubrutinib,Zanubrutinib
Indications:R/R FL
Clinical Trial:ROSEWOOD
Original Research Company: BeOne Medicines
Approval Date:2024.03.08
Approved Institution:FDA
On March 8, 2024, BeOne Medicines announced a BTK inhibitorZanubrutinibGranted FDA Accelerated Approval forCombination of anti-CD20 monoclonal antibody Obinutuzumab,Treatment of adult patients with R/R FL who have received at least two prior lines of systemic therapy. Since then, zanubrutinib has been approved for multiple indications overseas, including: Waldenström's macroglobulinemia (WM), adult mantle cell lymphoma (MCL), marginal zone lymphoma (MZL), CLL, and SLL.
This approval is based on a global, randomized, open-label Phase II clinical study.ROSEWOOD(NCT03332017)This study aims to evaluate the efficacy and safety of zanubrutinib in combination with obinutuzumab versus obinutuzumab monotherapy in patients with R/R FL. A total of 217 patients with R/R FL who had previously received second-line systemic therapy were enrolled. The results showed that, with a median follow-up time of 20 months, as assessed by the Independent Review Committee (IRC),The ORR was 69% in the zanubrutinib plus obinutuzumab group and 46% in the obinutuzumab group (P=0.0012).The combination of Zanubrutinib and Obinutuzumab demonstrated durable responses, with 69% of patients achieving a DOR of 18 months. The lasting and deep responses brought by the Zanubrutinib and Obinutuzumab combination are expected to translate into survival benefits.The median PFS of patients was significantly prolonged (28.0 months vs. 10.4 months).Moreover, the combination of zanubrutinib and obinutuzumab was generally well-tolerated, consistent with previous observations of these two drugs in prior studies.
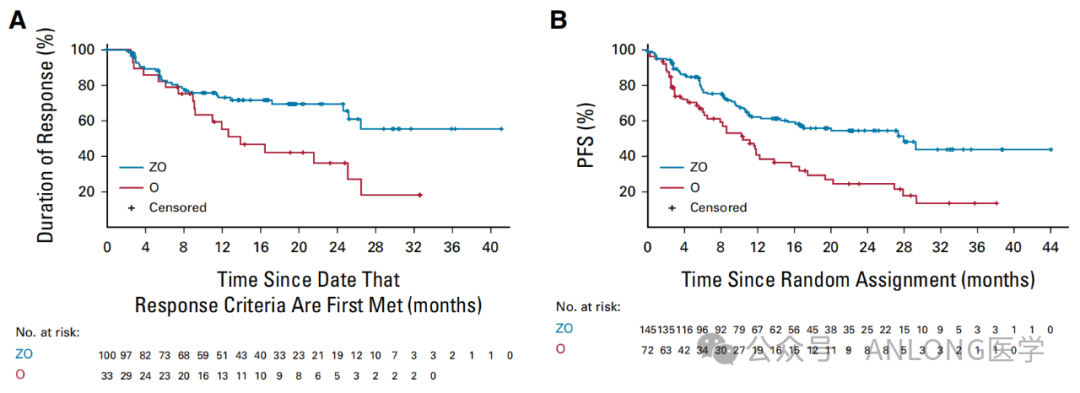
ROSEWOOD Study DOR and PFS Results
5. Breyanzi
Product Name:Breyanzi
Generic Name: lisocabtagene maraleucel, Relmacabtagene autoleucel
Indications: R/R CLL or SLL
Clinical Trial:TRANSCEND CLL 004
Original Research Company:Bristol-Myers Squibb
Approval Date:2024.03.14
Approved Institution:FDA
On March 14, 2024, Bristol-Myers Squibb announced that the FDA had granted accelerated approval.BreyanziUsed forAdult patients with R/R CLL or SLL who have received at least two prior treatments (including BTK inhibitors and BCL-2 inhibitors)Breyanzi is a CAR-T cell therapy targeting CD19, and it is also the first CAR-T cell therapy approved by the FDA for the treatment of CLL/SLL. It was first approved for marketing in the United States in February 2021, for the treatment of adult patients with relapsed or refractory large B-cell lymphoma (LBCL) who have received two or more systemic therapies.
This approval is based on an open-label, single-arm, multi-center Phase I/II clinical study.TRANSCEND CLL 004, aimed to evaluate the efficacy and safety of Breyanzi in R/R CLL or SLL patients. The results showed,20% of patients treated with Breyanzi achieved complete remission (CR).Among all responders (ORR of 45%), DOR was 35.3 months.Among patients treated with Breyanzi who achieved complete remission (CR), a high rate of minimal residual disease (MRD)-negativity was observed, with 100% MRD-negativity in blood and 92.3% MRD-negativity in bone marrow. No new safety signals were observed, and the incidence of cytokine release syndrome (CRS) and neurologic events (NEs) was mostly low-grade.
6. Toremifene
Product Name: Keloopin
Generic Name: Tolametinib, Tunlametinib
Indications: NRAS-Mutated Melanoma
Clinical Trial:NCT05217303
Original Research Company: Shanghai Kechow Pharma, Inc.
Approval Date:2024.03.15
Approved Institution:NMPA
On March 15, 2024, NMPA Approved MEK InhibitorTolamerinCapsule launched forPatients with advanced melanoma harboring NRAS mutations who have failed anti-PD-1/PD-L1 therapyToralisib is the first MEK inhibitor in China and the world's first and only targeted drug approved for the treatment of advanced melanoma with NRAS mutations. It can bind to MEK1/2 in the RAS/RAF/MEK/ERK signaling pathway, blocking the transmission of downstream signaling pathways, thereby inhibiting tumor growth.
This approval is based on the results of a single-arm, multi-center pivotal Phase II registration clinical study (NCT05217303), which aimed to evaluate the efficacy and safety of Toremifene in treating patients with advanced melanoma harboring NRAS mutations. The study enrolled a total of 100 patients with advanced NRAS-mutant melanoma, who were administered Toremifene orally at a dose of 12mg twice daily. Efficacy analysis was conducted on 95 patients. As of February 19, 2023, confirmed responses in the efficacy analysis set, as assessed by the IRC, showed thatORR was 34.7%, median PFS was 4.2 months, DCR was 72.6%, and the 1-year OS rate was 57.2%.The ORR was 39.1% in patients who had previously received immunotherapy. The most common treatment-related adverse events of grade 3 or higher included elevated blood creatine kinase, diarrhea, and edema, among others. Overall safety was manageable and controllable, and no participants died as a result of the study drug during the research process.
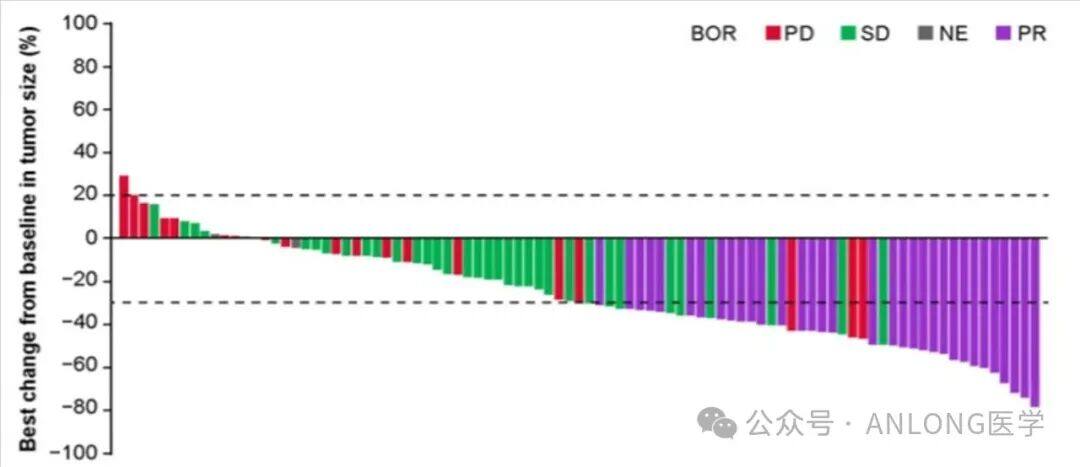
Waterfall Plot of Lesion Percentage Change
NRAS Gene MutationApproximately 1/4 of melanoma patients carry activating NRAS mutations.. This timeThe approval of Tauramis provides a new treatment option for melanoma patients with NRAS mutations.
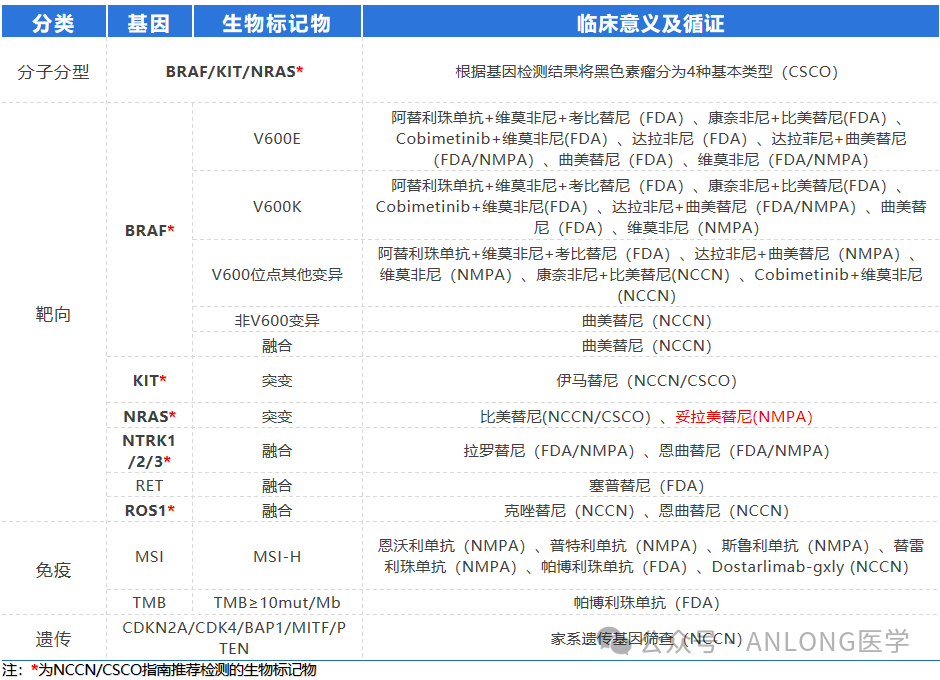
Summary of Recommended Biomarker Testing for Melanoma
The 2023 CSCO Guidelines for the Diagnosis and Treatment of Melanoma indicate that the evidence level for Tolorafib in the treatment of advanced melanoma has been elevated:
For patients with advanced cutaneous and acral melanoma (with or without brain metastasis) who carry the NRAS mutation, treatment with Toremifene is recommended. The recommendation has been elevated from a Level III second-line treatment to a Level I recommendation.
For patients with unresectable or stage IV mucosal melanoma who carry the NRAS mutation, treatment with Tolanib is recommended, moving from a previous Grade III recommendation to a Grade I recommendation.
7. Tislelizumab
Product Name: BeOne Medicines, Tevimbra
Generic NameTislelizumab
Indications:ESCC
Clinical Trial:RATIONALE 302
Original Research Company: BeOne Medicines
Approval Date:2024.03.15
Approved Institution:FDA
On March 15, 2024, BeOne Medicines announcedTislelizumabApproved by the FDA as a monotherapyAdult patients with unresectable or metastatic ESCC who have previously received systemic chemotherapy (excluding PD-1/L1 inhibitors)It is expected to be launched in the United States in the second half of 2024. This approval marks the first indication for tislelizumab in the United States.
This approval is based on a global, randomized, open-label Phase III clinical study.RATIONALE 302(NCT03430843)This study aims to compare the efficacy and safety of tislelizumab monotherapy with investigator's choice of chemotherapy as a second-line treatment for patients with unresectable, locally advanced or metastatic ESCC. A total of 513 patients from 132 research centers across 11 countries and regions in Europe, Asia, and North America were enrolled. The results showed that the study met its primary endpoint in the intention-to-treat (ITT) population.Compared with chemotherapy, tislelizumab demonstrated statistically significant and clinically meaningful survival benefits in the ITT population (median OS, 8.6 months vs. 6.3 months).Tislelizumab demonstrates a better safety profile compared to chemotherapy. The most common (≥20%) adverse reactions (including abnormal laboratory test results) are elevated glucose, decreased hemoglobin, decreased lymphocytes, and decreased sodium, among others.
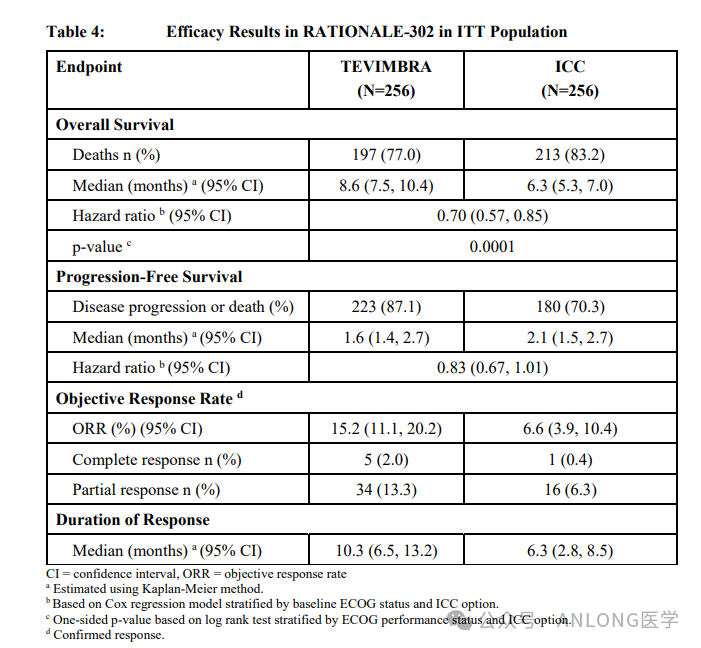
RATIONALE 302 Study Results
8. Sugrelimab
Product Name: CStone Pharmaceuticals
Generic Name: Sugemalimab, Sugemalimab
Indications: PD-L1 expression (CPS≥5)G/GEJ
Clinical Trial:GEMSTONE-303
Original Research CompanyCStone Pharmaceuticals
Approval Date:2024.03.15
Approved Institution:NMPA
On March 15, 2024, CStone Pharmaceuticals announced that NMPA approvedSugemalimabUnitedFluorouracil and Platinum-based ChemotherapyUsed forFirst-line treatment for PD-L1 expression (Combined Positive Score CPS≥5) in unresectable locally advanced or metastatic G/GEJ patients. This is the fifth indication approved by the NMPA for Sugemalimab, with the previous four indications being:
First-line combined chemotherapy for patients with metastatic squamous and non-squamous NSCLC;
Patients with unresectable, stage III NSCLC who have not experienced disease progression after concurrent or sequential chemoradiotherapy;
Treatment of patients with relapsed or refractory extranodal NK/T-cell lymphoma;
First-line treatment with fluorouracil and platinum-based chemotherapy for patients with unresectable locally advanced, recurrent, or metastatic ESCC.
This approval is based on a multicenter, randomized, double-blind, placebo-controlled Phase III clinical trial.GEMSTONE-303The study aims to evaluate the efficacy of Sugemalimab combined with Oxaliplatin and Capecitabine as a first-line treatment for unresectable PD-L1 expression (CPS).≥5) in locally advanced or metastatic G/GEJ. The primary endpoints of the study were investigator-assessed PFS and OS, with secondary endpoints including BICR-assessed PFS, investigator-assessed ORR, and DOR, among others. The study has met its pre-specified dual primary endpoints, and the results have been selected as a Late-Breaking Abstract (LBA) for the 2023 ESMO Congress, with detailed data presented in an oral report. The results show,The median PFS assessed by investigators in the Sugemalimab group vs. the placebo group was 7.6 months vs. 6.1 months, and the OS was 15.6 months vs. 12.6 months.Subgroup analysis showed that clinical benefits were observed across all predefined subgroups, including PD-L1 expression status. The ORR for the sugemalimab group vs. the placebo group was 68.6% vs. 52.7%, with a median DOR of 6.9 months vs. 4.6 months. Sugemalimab combined with chemotherapy demonstrated good tolerability and safety, with no new safety risks identified.
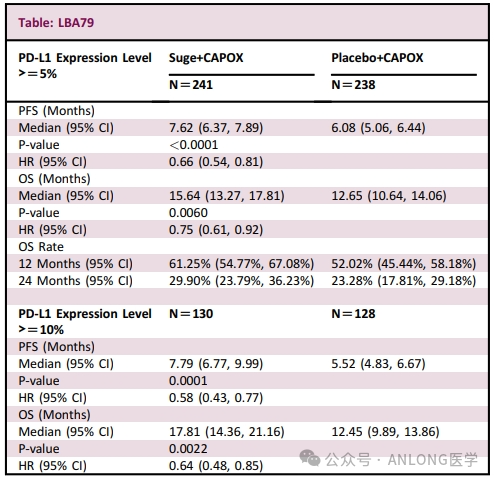
GEMSTONE-303 Study Results (from 2023 ESMO)
9. Ponatinib
Product Name:Iclusig
Generic NamePonatinib, Iclusig
Indications:Ph+ ALL
Clinical Trial:PhALLCON
Original Research Company:Takeda Pharmaceutical Company Limited
Approval Date:2024.03.19
Approved Institution:FDA
On March 19, 2024, the FDA granted accelerated approvalPonatinibUnitedChemotherapyUsed forTreatment of newly diagnosed Ph+ ALL adult patients.Following this approval, Ponatinib becomes the first and only targeted drug in the United States to be used in combination with chemotherapy as a first-line treatment for Ph+ ALL.
This approval is based on a randomized, active-controlled, open-label, multicenter Phase III clinical study.PhALLCON (NCT03589326)The study enrolled a total of 245 newly diagnosed adult patients with Ph+ ALL, who were randomly assigned in a 2:1 ratio to receive either Ponatinib combined with chemotherapy or Imatinib combined with chemotherapy. The chemotherapy regimen included three cycles of induction therapy with Vincristine and Dexamethasone, six cycles of alternating consolidation therapy with Methotrexate and Cytarabine, and 11 cycles of maintenance therapy with Vincristine and Prednisone. The primary endpoint was the CR rate with negative MRD. The results showed,In the ponatinib group and imatinib group, the MRD-negative CR rates at the end of induction were 30% and 12%, respectively.
Ponatinib is a third-generation TKI that targets BCR-ABL1 and is active against wild-type and BCR-ABL1 mutant diseases, including the T315I mutation.For patients with chronic myeloid leukemia (CML) in chronic phase (CP), accelerated phase (AP), or blast phase (BP) who are resistant or intolerant, as well as for Ph+ ALL or T315I mutation-positive patients.
10. Mirvetuximab soravtansine-gynx
Product Name:Elahere
Generic Name:Mirvetuximab soravtansine-gynx
Indications:FRα-positiveOvarian Cancer, Fallopian Tube Cancer, or Primary Peritoneal Cancer
Clinical Trial:MIRASOL
Original Research Company: AbbVie
Approval Date:2024.03.22
Approved Institution:FDA
On March 22, 2024, AbbVie announced that the FDA had fully approvedMirvetuximab soravtansine-gynxUsed forTreatmentPatients with FRα-positive, platinum-resistant epithelial ovarian cancer, fallopian tube cancer, or primary peritoneal cancer who have previously received 1-3 lines of systemic therapy.Previously, on November 15, 2022, the product had received FDA accelerated approval for marketing.
This full approval is based on an open-label, multi-center, positive drug-controlled, randomized Phase III clinical study.MIRASOL(NCT04209855)The study enrolled a total of 453 patients with FRα-positive (IHC, ≥75%, 2+/3+) platinum-resistant epithelial ovarian cancer, fallopian tube cancer, or primary peritoneal cancer who had previously received 1-3 lines of systemic therapy. The trial recruited patients determined to have positive tumor FRα expression by the VENTANA FOLR1 (FOLR1-2.1) RxDx assay. Patients were randomly assigned 1:1 to receive either Mirvetuximab soravtansine-gynx (n=227) or investigator’s choice chemotherapy (paclitaxel, pegylated liposomal doxorubicin, or topotecan, n=226) until disease progression or unacceptable toxicity occurred. The results showed,The OS for the Mirvetuximab soravtansine-gynx group and the chemotherapy group were 16.5 months and 12.7 months, respectively, with median PFS of 5.6 months and 4.0 months, and ORR of 42% and 16%, respectively.Regarding safety, 54% and 34% of patients required dose interruptions and reductions of Elahere due to adverse events (AEs), respectively, and 9% of patients permanently discontinued Elahere treatment due to AEs. The most common toxicities leading to discontinuation included pneumonia (2%), blurred vision (1%), and peripheral neuropathy (1%). Serious AEs occurred in 24% of patients.
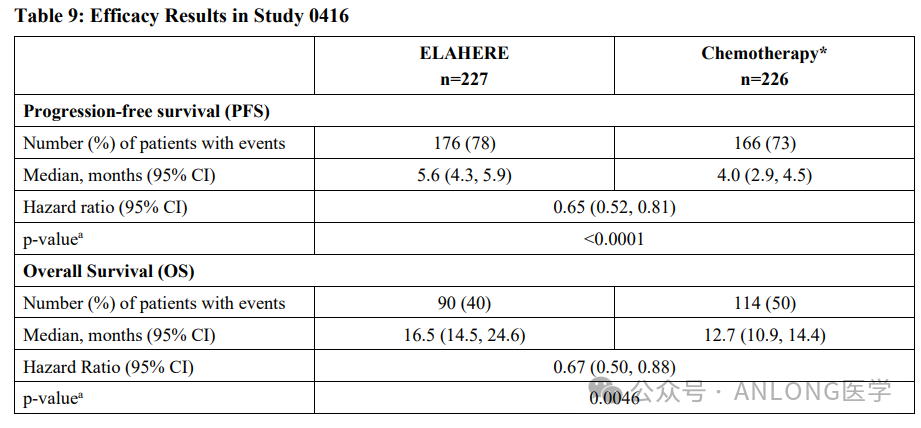
MIRASOL Study Results
FRα is expressed in non-malignant tissues such as choroid plexus, thyroid, and breast, with limited distribution, but is overexpressed in various epithelial tumors, particularly ovarian cancer, endometrial cancer, and triple-negative breast cancer. Based on its highly tumor-restricted expression profile, FRα represents an attractive therapeutic target for ovarian cancer. Mirvetuximab soravtansine-gynx is an ADC drug targeting FRα, which binds with high affinity to FRα expressed on the surface of tumor cells, promoting the internalization of the ADC/receptor complex through antigen-mediated endocytosis. Lysosomal processing releases the active DM4 metabolite—a maytansine derivative—which inhibits tubulin polymerization and microtubule assembly, inducing potent anti-mitotic effects that lead to cell cycle arrest and apoptosis. The active metabolite may also diffuse into neighboring cells, inducing further cell death (referred to as the "bystander effect"). CurrentlyMirvetuximab soravtansine-gynxIt has been included in the 2024.v1 edition of the NCCN Guidelines for Ovarian Cancer/Fallopian Tube Cancer/Primary Peritoneal Cancer, with or without bevacizumab, for the treatment of FRα-positive platinum-resistant or platinum-sensitive ovarian cancer/fallopian tube cancer/primary peritoneal cancer.

2024.v1 NCCN Guidelines for Ovarian Cancer/Fallopian Tube Cancer/Primary Peritoneal Cancer (Treatment for Platinum-Sensitive Recurrent Disease)

2024.v1 NCCN Guidelines for Ovarian Cancer/Fallopian Tube Cancer/Primary Peritoneal Cancer (Treatment for Platinum-Resistant Recurrent Disease)
11. Zolbetuximab
Product Name:Vyloy
Generic Name:Zolbetuximab
IndicationsCLDN18.2-positive gastric cancer
Clinical Trial:SPOTLIGHT、GLOW
Original Research Company: Astellas
Approval Date:2024.03.26
Approved Institution:MHLW
On March 26, 2024, Astellas announced the approval by MHLW.ZolbetuximabUsed forTreatment of CLDN18.2-positive, unresectable, advanced or recurrent gastric cancer patients.Zolbetuximab Becomes the World's First and Only Approved CLDN18.2-Targeted Therapy.
This approval is mainly based onSPOTLIGHT(NCT03504397)AndGLOW(NCT03653507)Positive results from the Phase III clinical trials, which were used to evaluate the efficacy and safety of first-line treatment in CLDN18.2-positive, HER2-negative locally advanced or metastatic G/GEJ adenocarcinoma patients. SPOTLIGHT evaluated the efficacy comparison between Zolbetuximab + mFOLFOX6 (a combination chemotherapy regimen including oxaliplatin, leucovorin, and fluorouracil) and placebo + mFOLFOX6. The GLOW study assessed the efficacy comparison between Zolbetuximab + CAPOX (a combination chemotherapy regimen including capecitabine and oxaliplatin) and placebo + CAPOX. The results showed:
SPOTLIGHT Study: Compared with the placebo group, the PFS and OS in the Zolbetuximab group showed statistically significant improvement.The PFS for the Zolbetuximab group and placebo group were 10.61 months and 8.67 months, respectively, while the OS was 18.23 months and 15.54 months, respectively.The incidence rates of severe treatment-related adverse reactions were similar between the two groups, at 44.8% and 43.5%, respectively, consistent with previous trial results.
GLOW Study: Compared with the placebo group, the PFS in the Zolbetuximab group showed a statistically significant improvement.The PFS for the Zolbetuximab group and placebo group were 8.21 months and 6.8 months, respectively, while the OS was 14.39 months and 12.16 months, respectively.
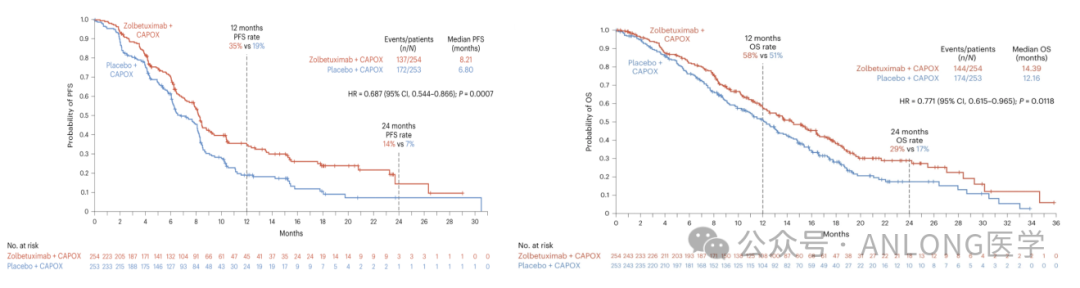
GLOW Study PFS and OS Results
The expression rate of Claudin18.2 protein is 40-80% in gastric cancer patients and 50-70% in pancreatic cancer patients.In addition, Claudin18.2 can be abnormally expressed in various primary malignant tumors such as breast cancer, colorectal cancer, biliary tract tumors, head and neck cancer, and non-small cell lung cancer. Its highly selective expression distribution and extremely high positive rate across multiple cancer types make it a potential pan-cancer target for anti-tumor drugs.
Currently,Claudin18.2 has been included in the 2023 edition of the CSCO Gastric Cancer Guidelines.Claudin18.2 IHC Testing is Listed as a Level III Recommendation for Molecular Testing in Metastatic Gastric Cancer PatientsClass 2B),For patients with advanced or recurrent gastric cancer who have failed standard treatment, biomarker tests such as Claudin 18.2, FGFR2, C-MET, and NTRK genes can be performed to identify potential therapeutic targets.

2023 Edition of the CSCO Gastric Cancer Guidelines (Molecular Diagnostics)
Zolbetuximab is an IgG1 monoclonal antibody that specifically binds to CLDN18.2 on the surface of tumor cells to exert its anti-tumor effects. Preclinical studies have shown that this binding induces cancer cell death through the activation of two distinct immune system pathways—antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC)—to achieve the purpose of tumor treatment.
12. Idecabtagene Vicleucel
Product Name:Fukasu
Generic Name:Idecabtagene Vicleucel
Indications:R/R MM
Clinical Trial:FUMANBA-1
Original Research Company: IASO Bio
Approval Date:2024.03.28
Approved Institution:NMPA
On March 28, 2024, IASO Bio announcedIdecabtagene VicleucelApproved by NMPA, Indication Expansion PlannedFor the treatment of R/R MM patients who have previously undergone 1-2 lines of therapy and are lenalidomide-resistant.Equecabtagene Autoleucel Injection is a CAR-T product targeting BCMA. It was previously approved for marketing in China in June 2023, for the treatment of R/R MM patients who have progressed after receiving at least three prior lines of therapy (including at least one proteasome inhibitor and one immunomodulatory agent).
This approval is based on a multi-center Phase I/II clinical study conducted in China.FUMANBA-1(CTR20192510, NCT05066646)According to the latest long-term follow-up data from this study presented at the 2023 International Myeloma Society (IMS) Annual Meeting, as of December 31, 2022, among 103 evaluable participants,The ORR was 96.1%, and the stringent complete response/complete response (sCR/CR) rate was 77.7%. Among the 91 subjects without a history of prior CAR-T therapy, the ORR reached 98.9%, the sCR/CR rate reached 82.4%, and the 12-month PFS rate was 85.5%.94.2% (97/103) of subjects achieved MRD negativity, and all CR/sCR subjects reached MRD negativity. Among the 105 treated subjects, only one subject experienced ≥Grade 3 cytokine release syndrome (CRS), with no instances of ≥Grade 3 immune effector cell-associated neurotoxicity syndrome (ICANS).
More comprehensive information on the latest tumor treatment drugs recently launched in China and abroad can be found below.
New Drug Express | Overview of Anti-Tumor Drug Approvals in China and Globally in November 2023
New Drug Express | Overview of Anti-Tumor Drug Approvals in China and Globally (July-August 2023)
Anlong Gene

Anlong Gene Technology Co., Ltd. (referred to as Anlong Gene) was established on September 1, 2016. The founding team members are from the School of Medicine at Tsinghua University. It is a national high-tech enterprise that simultaneously provides third-party testing services and engages in the research, development, production, registration, and sales of IVD reagents and instruments.
The company's R&D technology focuses on the convenience of clinical application, featuring simpler operations and faster processes. It has developed several precision oncology companion diagnostic/auxiliary treatment technologies, including the OmegaSeq amplicon capture library construction technology, multiplex primer/probe dual-block ctDNA methylation detection technology, and automated data analysis technology. The company has established a cancer gene database comprising over 100,000 cases across nearly 30 types of cancers, forming a technical barrier within the industry.
In addition, the company strictly follows the ISO15189 standard to establish a quality management system for medical laboratories, comprehensively improving the service quality of the laboratory and ensuring the impartiality, scientific nature, and accuracy of test reports. The laboratory has built a variety of testing platforms, including PCR, digital PCR, first-generation sequencing, and second-generation sequencing. Its third-party testing service products cover the entire cycle of services such as precise immunotherapy, tumor precision medication guidance, early tumor screening, hereditary tumor screening, and prognosis prediction, providing an overall solution for precise tumor diagnosis and treatment. The company owns several domestic pioneering cancer early screening products and has developed, for clinical use, an automatic nucleic acid extraction vulcanizer with independent intellectual property rights and as a domestic first in China.
Currently, Anlong Gene has achieved full marks in multiple China clinical laboratory quality evaluations and has received several honorary titles, including "National High-tech Enterprise," "Anhui Province Strategic Emerging Industry Cluster Development Base," "Anhui Province High-level Science and Technology Talent Team," and "Top 500 Hidden Unicorn Enterprises in China."
Anlong Gene

Anlong Medicine
