AstraZeneca Showcases Multiple Clinical Advances in Gastrointestinal Cancers at ASCO 2024

AstraZeneca
Biopharmaceutical Manufacturer
*For medical professionals only
Frontier Pursuit: A Comprehensive Overview.
Recently, the 2024 American Society of Clinical Oncology (ASCO) Annual Meeting was grandly held in Chicago, USA, from May 31 to June 4, Central Daylight Time. In this conference, significant breakthroughs were achieved in multiple Phase I, II, and III clinical studies in the field of gastrointestinal tumors. These include immunotherapy drugs such as Durvalumab and Tremelimumab, targeted therapy drugs like Trastuzumab Deruxtecan targeting HER2, AZD0901 targeting Claudin 18.2, and innovative CAR-T therapies such as C-CAR031. These studies not only guide new methods and directions for the treatment of gastrointestinal tumors but also bring new hope to a large number of patients with gastrointestinal tumors. The editor has specially selected parts of the research content and compiled them into an article for readers' reference.
Abstract No.: 4122
Safety Analysis of Each Treatment Phase in the EMERALD-1 Study: A Randomized, Placebo-Controlled Phase III Trial of Transcatheter Hepatic Artery Chemoembolization Combined with Durvalumab and/or Bevacizumab for Unresectable Hepatocellular Carcinoma Suitable for Embolization
Background:Primary Endpoint of Global Phase III EMERALD-1 Study Met: In Patients with Unresectable Hepatocellular Carcinoma (uHCC) Suitable for Embolization, Durvalumab (D) + Bevacizumab (B) + TACE Significantly Improved Progression-Free Survival (PFS) [Median 15.0 Months vs 8.2 Months; HR=0.77; 95% CI: 0.61-0.98; p=0.032 (Threshold 0.0435)] Compared to Placebo + TACE, with Manageable Safety. This Study Conducted a Post Hoc Exploratory Analysis of Safety in Two Treatment Phases [D-TACE (D-T) and D-B].
Method:Patients were randomly assigned in a 1:1:1 ratio to the D+TACE, D+B+TACE, or placebo+TACE groups. During the D-T treatment phase, patients received 1-4 sessions of TACE [cTACE or DEB-TACE (investigator’s choice)] plus D (1500mg once every 4 weeks) or placebo for D. During the D-B treatment phase, after the last TACE, patients received D (1120mg once every 3 weeks) plus placebo for B, or D (1120mg once every 3 weeks) plus B (15mg/kg once every 3 weeks), or placebo for D + placebo for B. For patients who received any study treatment and met randomization criteria, adverse events (AEs), start and end dates of AEs, maximum CTCAE grade/changes, and causality were assessed during these two treatment phases (D-T and D-B) until the end of follow-up.
Results:The table shows the duration of exposure (Table 1), with the number (%) of patients experiencing AEs in the D+TACE, D+B+TACE, and placebo+TACE groups during the D-T treatment phase being 144 cases (74.6%), 139 cases (72.0%), and 148 cases (74.0%), respectively. During the D-B treatment phase, the respective numbers were 133 cases (68.9%), 147 cases (76.2%), and 132 cases (66.0%). The number (%) of patients with AEs potentially related to study treatment during the D-T treatment phase were 59 cases (30.6%), 56 cases (29.0%), and 41 cases (20.5%), while during the D-B treatment phase, the respective numbers were 76 cases (39.4%), 114 cases (59.1%), and 69 cases (34.5%). The number (%) of patients with AEs triggered by TACE during the D-T treatment phase were 72 cases (37.3%), 90 cases (46.6%), and 85 cases (42.5%), while during the D-B treatment phase, the respective numbers were 16 cases (8.3%), 18 cases (9.3%), and 21 cases (10.5%).
Conclusion:During the D-T and D-B treatment phases, D+B+TACE demonstrated controllable safety, consistent with the safety signals of monotherapy and underlying diseases. These data further support D+B+TACE as a new potential standard treatment option for uHCC patients suitable for embolization.
Clinical trial information: NCT03778957
Table 1 Safety Data Related to Different Treatment Groups
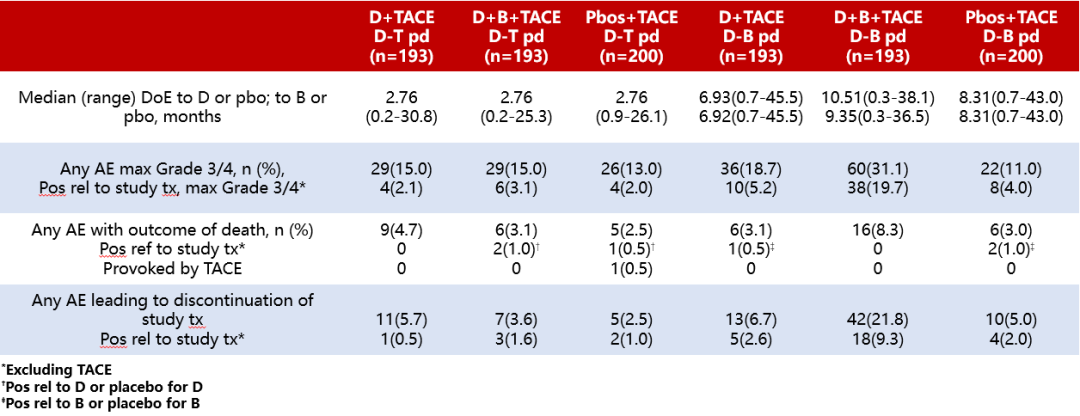
Note: DoE is the duration of exposure.
Abstract No.: 4090
Trastuzumab Deruxtecan (T-DXd) in HER2-Expressing Biliary Tract Cancer (BTC) and Pancreatic Cancer (PC) Patients: Results from the DESTINY-PanTumor02 (DP-02) Study.
Background:Currently, the long-term benefits of later-line treatment options for BTC and PC are quite limited. In the DP-02 study, T-DXd demonstrated therapeutic potential across various solid tumors, with an objective response rate (ORR) of 37.1% (95% CI: 31.3-43.2) in 267 patients with HER2-expressing tumors, achieving clinically meaningful survival outcomes. Here, we report the subgroup analysis of the BTC and PC cohorts and describe the characteristics of patients with objective responses (OR).
Method:This open-label Phase II clinical trial evaluated the efficacy of T-DXd (5.4 mg/kg, once every 3 weeks) in patients with locally advanced or metastatic solid tumors who were either previously treated (≥1 line) or untreated and had HER2 expression [immunohistochemistry (IHC) 3+ or 2+] confirmed by local or central laboratory testing. The primary endpoint was investigator-assessed ORR. Secondary endpoints included PFS and safety. Exploratory endpoints encompassed efficacy outcomes based on HER2 expression levels.
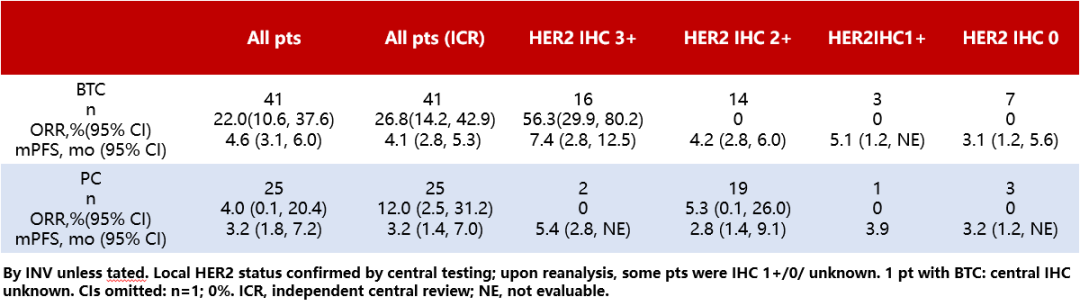
Results:The results showed that, at the data cutoff (June 2023), 41 BTC patients and 25 PC patients had received T-DXd treatment [median follow-up durations were 6.01 months (range: 0.7-29.1 months) and 4.99 months (range: 1.1-27.2 months), respectively]. Among BTC and PC patients, 27 (65.9%) and 18 (72.0%) had previously received ≥2 lines of therapy, 7 (17.1%) and 2 (8.0%) had prior anti-HER2 therapy, and 8 (19.5%) and 18 (72.0%) had prior topoisomerase 1 (TOP1) inhibitor therapy, respectively. Further analysis of efficacy in all patients and those with different HER2 expression levels (Table 2) revealed that among BTC patients with HER2 expression (IHC 3+), 9 patients (56.3%; 95% CI: 29.9-80.2; primary tumor locations: ampulla, 2; extrahepatic, 2; gallbladder, 4; intrahepatic, 1) were confirmed by investigators to have OR. Of these patients, 6 had received ≥2 prior lines of therapy, and 4 had PD-L1 expression ≥1%. Among BTC and PC patients, drug-related ≥grade 3 AEs occurred in 16/41 (39.0%) and 7/25 (28.0%), respectively; confirmed drug-related interstitial lung disease or pneumonia occurred in 7/41 (17.1%; grade 2, 5; grade 3, 1; grade 5, 1) and 1/25 (4.0%; grade 1, 1), respectively.
Conclusion:In summary, T-DXd has demonstrated clinical significance in BTC patients with different primary tumor sites. Although the number of patients in the PC cohort is relatively small, which imposes certain limitations on this result, these data still support further exploration of the application value of T-DXd in PC patients. Meanwhile, its safety profile is consistent with previously known information. These data will support T-DXd as a potential treatment option for HER2-expressing BTC patients.
Clinical trial information: NCT04482309
Table 2 Efficacy in All Patients and in Patients Stratified by HER2 Expression Levels
Abstract No.: 4019
C-CAR031, a TGFβRIIDN-armored CAR-T therapy targeting GPC3, in a Phase I study for advanced hepatocellular carcinoma (HCC) patients
Background:Autologous anti-glypican-3 (GPC3), a surface antigen overexpressed in hepatocellular carcinoma (HCC) but nearly absent in healthy tissues, offers a promising new therapeutic option for advanced unresectable HCC through autologous chimeric antigen receptor T cells (CAR-T) targeting GPC3. C-CAR031 is an autologous GPC3-directed CAR-T armored with a dominant-negative TGF-β receptor II (TGFβRIIDN). This study reports the safety and preliminary efficacy of C-CAR031 in patients with advanced HCC.
Method:This study is a first-in-human, open-label Phase I clinical trial using a dose-escalation design that combines accelerated titration with the i3+3 method. GPC3+ patients with advanced HCC who had previously failed ≥1 line of systemic treatment regimens received a single intravenous injection of C-CAR031 after standard lymphodepletion therapy. The primary endpoints were safety and tolerability, with other endpoints including pharmacokinetics and preliminary efficacy. Adverse events (AEs) were graded according to CTCAE 5.0, and cytokine release syndrome (CRS)/immune effector cell-associated neurotoxicity syndrome (ICANS) were assessed based on ASTCT 2019 criteria. Investigators evaluated objective responses according to RECIST v1.1.
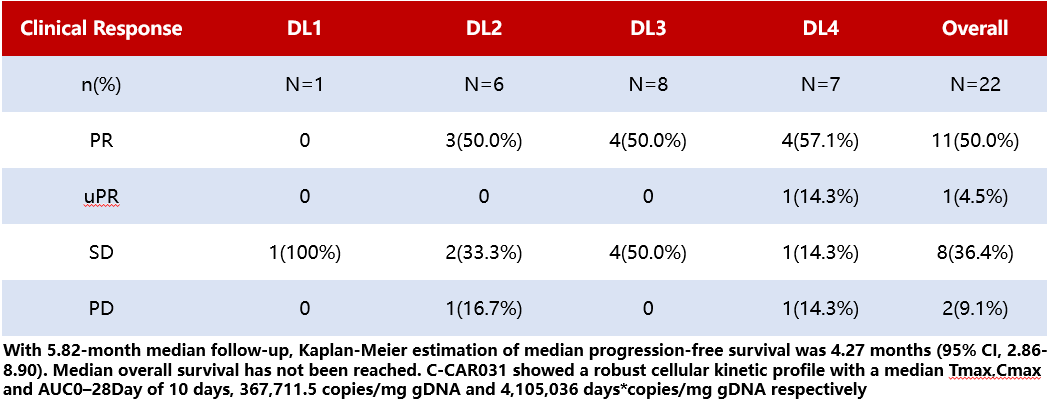
Results:The results (Table 3) showed that, as of January 5, 2024, a total of 24 patients received C-CAR031 injections across four dose levels (DL1-DL4). All patients had BCLC-C stage HCC, and 20/24 cases (83.3%) exhibited extrahepatic metastases. The median number of prior treatment lines was 3.5 (range 1-6), with 23 patients (95.8%) having received immune checkpoint inhibitors (ICIs) and tyrosine kinase inhibitors (TKIs). Safety was evaluable in all patients. No dose-limiting toxicity or ICANS was observed. Among the 22 patients (91.7%) who experienced CRS, only one case (4.2%) was grade 3. The most common ≥grade 3 AEs were lymphopenia (100%), neutropenia (70.8%), thrombocytopenia (37.5%), and elevated transaminases (16.7%). One patient (4.2%) developed grade 4 bone marrow suppression, and one patient (4.2%) at DL4 developed grade 3 interstitial pneumonia due to grade 3 CRS. All AEs were reversible. Efficacy was evaluable in 22 patients. Tumor shrinkage was observed in both intrahepatic and extrahepatic lesions in 90.9% of patients, with a median reduction rate of 44.0% (range 3.4%-94.4%). Across all patients (DL1-DL4), the disease control rate was 90.9%, and the ORR was 50.0%. At DL4, the ORR was 57.1%.
Conclusion:This study shows that C-CAR031 demonstrates manageable safety and encouraging anti-tumor activity in heavily treated advanced HCC patients.
Clinical trial information: NCT05155189
Table 3 Efficacy by Injection Dose Stratification (DL1-DL4) and All Patients
Abstract No.: 4022
Pharmacodynamic Study of T-cell Receptor and Immune Gene Expression in Patients with uHCC Treated with Durvalumab Alone or in Combination with Tremelimumab or Bevacizumab
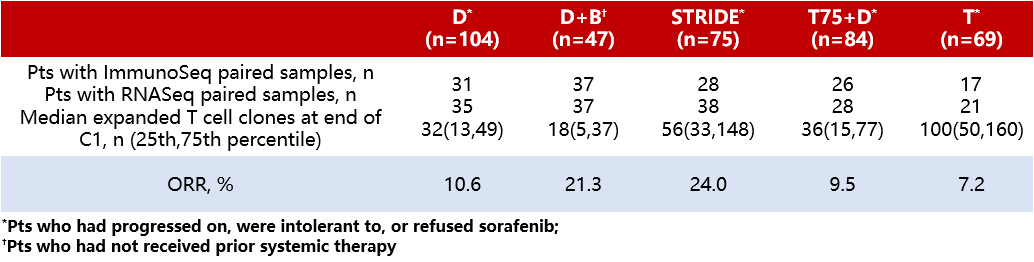
Background:Study22 (NCT02519348) is a Phase II clinical trial evaluating immune checkpoint inhibitor (ICI) monotherapy and combination therapies for uHCC. It has been shown that the ORR is higher with the STRIDE regimen (a single dose of tremelimumab (T) + regular interval dosing of durvalumab (D)) or the D + bevacizumab (B) regimen compared to durvalumab (D) monotherapy [1, 2]. Previous pharmacodynamic analyses have indicated that the STRIDE regimen improves efficacy over D monotherapy, which is associated with increased T-cell clonal expansion by the end of the first cycle (C1) [3]. Therefore, this study further analyzed T-cell clones, including those in the D + B regimen, and compared the gene expression signatures (GES) across treatment groups to explore the similarities and differences in biomarker mechanisms underlying the efficacy of the STRIDE and D + B regimens.
Method:Patients with uHCC who had not previously received ICI treatment were randomly assigned to five regimen groups: the STRIDE regimen [T 300mg + D 1500mg, followed by D 1500mg (Q4W) alone after one cycle]; the T75+D regimen [T 75mg + D 1500mg, followed by sequential D 1500mg (Q4W) after four cycles]; the D monotherapy regimen [D 1500mg (Q4W)]; the T monotherapy regimen [T 750 mg, Q4W for the first seven cycles, then Q12W after seven cycles]; or the D+B regimen [D 1120mg + B 15mg/kg (Q3W)]. DNA and RNA were isolated from whole blood samples collected at baseline and at the end of the first cycle, which were preserved using PAXgene. The CDR3 region of the T-cell receptor β chain was analyzed using the immunoSEQ Assay (an immune sequencing platform developed by Adaptive Biotechnologies in Seattle, USA), and RNA sequencing was performed (supported by QSquared Solutions in North Carolina, USA). This study reports the relationship between the sequencing results from the ImmunoSeq platform and changes in immune gene expression with ORR.
Results:The sequencing results of the ImmunoSeq platform did not indicate significant differences in baseline T-cell clonality among different treatment groups. Compared with the D monotherapy regimen, an increase in T-cell clonal expansion was observed at the end of the first cycle with the STRIDE regimen, whereas no change in the number of T-cell clonal expansions was observed with the D+B regimen compared to the D monotherapy regimen. No association was observed between T-cell clonal expansion and ORR in this analysis (Table 4). Additionally, further analysis of GES showed that although upregulated expression of Type I and Type II interferons and mitosis-related genes was observed in both the STRIDE and D+B regimens, the changes in immune-related peripheral GES were more pronounced with the STRIDE regimen. In particular, this study observed noticeable changes in the GES of CD8+ T cells, M1 macrophages, and regulatory T cells (Tregs) only in the STRIDE regimen.
Conclusion:Although both the STRIDE and D+B regimens showed higher ORR in uHCC patients compared to the D monotherapy regimen, an increase in T-cell clonal expansion was only observed with the STRIDE regimen versus the D monotherapy. Furthermore, differences in the magnitude and type of GES changes suggest that the STRIDE and D+B regimens have distinct and potentially complementary mechanisms of action.
References:1. Kelley, et al. J Clin Oncol 2021. 2. Lim, et al. J Clin Oncol2022, A436. 3. McCoon, et al. J Clin Oncol2021, A4087
Clinical trial information: NCT02519348
Table 4 Median T-cell Clonal Expansion and ORR at the End of the First Cycle for Different Treatment Regimens
Abstract No.: TPS4199
ARTEMIDE-Biliary01: A Phase III Randomized Trial of Rilvegostomig Combined with Chemotherapy as Adjuvant Treatment for Resected Biliary Tract Tumors
Background:Biliary Tract Cancer (BTC) is a rare and aggressive heterogeneous gastrointestinal tumor that occurs in the intrahepatic or extrahepatic bile ducts [cholangiocarcinoma (CCA)] or gallbladder cancer (GBC). For early-stage patients who have undergone radical surgery, adjuvant chemotherapy based on fluoropyrimidine or gemcitabine is usually recommended, but the recurrence rate remains high (e.g., in the BILCAP study, the 5-year recurrence-free survival rate with capecitabine as adjuvant chemotherapy was 34%). Given the efficacy of immunotherapy in the adjuvant treatment of other cancer types, the results of the TOPAZ-1 and KEYNOTE-966 studies support the combination of immunotherapy and chemotherapy for advanced BTC (including locally advanced non-metastatic disease). Additionally, inhibitors targeting PD-1 or PD-L1, as well as novel immune checkpoint inhibitors such as TIGHT inhibitors (T-cell immunoglobulin and ITIM domain), have shown promising effects across various tumor types without a significant increase in high-grade toxicity compared to inhibiting PD-1 or PD-L1 alone. Rilvegostomig, an anti-PD-1/-TIGIT bispecific monoclonal antibody, has demonstrated favorable antitumor activity and tolerability in NSCLC. The ARTEMIDE-Biliary01 study will explore the efficacy of rilvegostomig combined with chemotherapy as adjuvant therapy for BTC patients after radical resection.
Method:ARTEMIDE-Biliary01 (NCT06109779) is a Phase III, randomized, double-blind, placebo-controlled, multicenter, global study designed to evaluate the efficacy and tolerability of rilvegostomig IV, Q3W, compared to placebo, in combination with investigator’s choice of chemotherapy [capecitabine, S-1 (tegafur/gimeracil/oteracil), or gemcitabine/cisplatin] as adjuvant treatment for patients with BTC after radical resection. The study will enroll approximately 750 adult patients histologically confirmed with biliary adenocarcinoma (intrahepatic or extrahepatic CCA or muscular invasive GBC), who have undergone macroscopically complete resection (R0 or R1) and have an ECOG performance status of 0-1. Key exclusion criteria include locally advanced, unresectable, or metastatic disease at initial diagnosis, and prior anti-BTC therapy before surgery. The primary endpoint is recurrence-free survival (RFS), with overall survival (OS) as a key secondary endpoint. Other endpoints include post-recurrence PFS, patient-reported tolerability, and safety. Recruitment for the study has begun, with approximately 200 research sites across Asia, Australia, Europe, North America, and South America participating globally.
Clinical trial information: NCT06109779
Abstract Number: TPS4187
GEMINI-Hepatobiliary: A Phase II Study of First-Line Treatment with a Novel Immunotherapy for Patients with Advanced Hepatobiliary Tumors
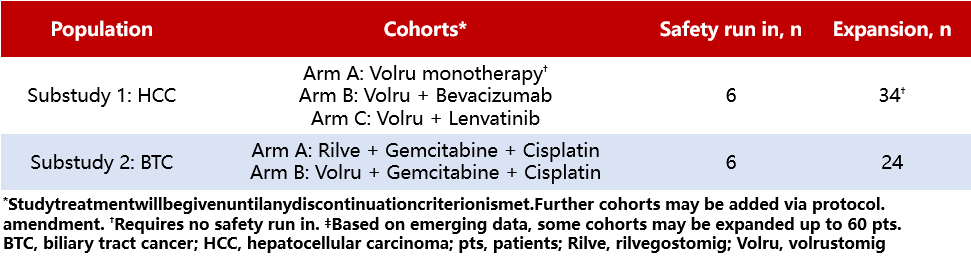
Background:Despite the improvement in overall survival (OS) for advanced hepatobiliary tumors with targeted therapies and immune-based (IO) treatment regimens, long-term survival rates remain unsatisfactory. Previous studies have shown that volrustomig (volru, a PD-1/CTLA-4 bispecific humanized IgG1 monoclonal antibody) is effective in patients with non-small cell lung cancer (NSCLC) and renal cell carcinoma, and rilvegostomig (rilve, a PD-1/TIGIT bispecific humanized IgG1 antibody) has demonstrated efficacy in NSCLC patients, with both showing manageable safety profiles. GEMINI-Hepatobiliary (NCT05775159) is a Phase II, open-label, multi-drug, multi-center, master protocol clinical trial designed to evaluate the preliminary efficacy and safety of novel IO bispecific antibodies as monotherapy or in combination with standard anti-cancer agents for patients with advanced hepatocellular carcinoma (HCC) and biliary tract cancer (BTC).
Method:GEMINI-Hepatobiliary Platform Design Allows for Independent Sub-Studies (Table 5). Sub-Study 1 includes adult patients (≥18 years) with previously untreated, histologically confirmed advanced HCC, ineligible for locoregional therapy, Barcelona Clinic Liver Cancer (BCLC) stage B/C, Child-Pugh class A score 5-6, and ECOG performance status (PS) of 0-1. Enrolled patients will receive volru monotherapy or in combination with bevacizumab/lenvatinib. The primary endpoints are safety and investigator-assessed objective response rate (ORR) per RECIST v1.1. Sub-Study 2 enrolls previously untreated adult patients with histologically confirmed locally advanced or metastatic BTC and an ECOG score of 0-1. Enrolled patients will receive gemcitabine + cisplatin in combination with rilve or volru. The primary endpoints are safety and investigator-assessed progression-free survival (PFS) per RECIST v1.1. Secondary endpoints for both sub-studies include duration of response (DOR), disease control rate (DCR), ORR, PFS, overall survival (OS), pharmacokinetics, and immunogenicity. Although no formal statistical comparisons or hypothesis testing is planned, the sample size was chosen to provide adequate precision for the primary endpoints. The study is currently recruiting at sites in the United States, Asia, and Europe.
Clinical trial information: NCT05775159
Table 5 HCC and BTC Sub-studies
Abstract No.: 434420
Phase I Clinical Study of Claudin18.2-Specific Antibody-Drug Conjugate CMG901 in Patients with Advanced Gastric Cancer/Gastroesophageal Junction Adenocarcinoma
Background:Claudin18.2 is a promising therapeutic target for advanced gastric cancer/gastroesophageal junction adenocarcinoma (G/GEJ). CMG901 is a potential first-in-class antibody-drug conjugate targeting Claudin18.2, conjugated with the payload monomethyl auristatin E (MMAE) via a linker, and has demonstrated potent anti-tumor activity in preclinical studies.
Method:This Phase I clinical trial includes a dose-escalation phase (Group A; 0.3-3.4 mg/kg) and a dose-expansion phase (Group B; 2.2, 2.6, and 3.0 mg/kg) to evaluate the safety, tolerability, and anti-tumor activity of CMG901 in patients with G/GEJ and other solid tumors. Patients enrolled in Group A are not required to have CLDN18.2 expression, whereas G/GEJ patients in Group B must meet specific criteria: at least 5% (i.e., ≥5%) of tumor cells must exhibit membrane staining intensity of at least 2+ (i.e., ≥2+) for CLDN18.2 expression to be eligible for the study. Patients receive CMG901 via intravenous infusion, Q3W, until disease progression or the occurrence of unacceptable toxicity. The primary endpoints for Group A are safety, tolerability, and the determination of the maximum tolerated dose (MTD), while the primary endpoints for Group B are ORR (according to RECIST v1.1 criteria) and the recommended Phase II dose. This study is ongoing, and the relevant data for G/GEJ patients are presented below.
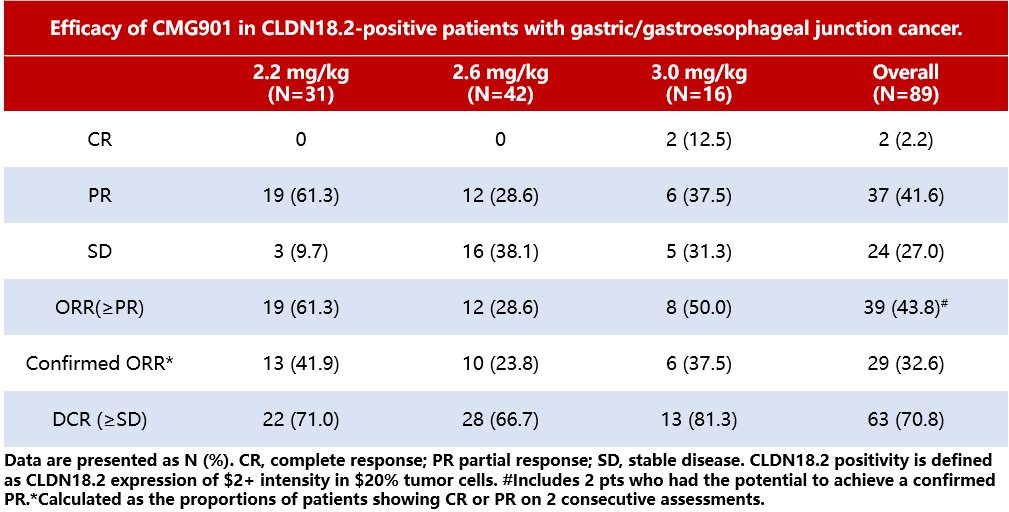
Results:The MTD was not reached during the dose escalation period. As of July 24, 2023, a total of 113 G/GEJ patients received CMG901 treatment at doses ranging from 2.2-3.0mg/kg (6 in Group A, 107 in Group B), with a median of 2 prior lines of systemic therapy (range 1-6). The most common treatment-emergent adverse events (TEAEs) were anemia (62.8%), vomiting (57.5%), and hypoalbuminemia (57.5%). The most common ≥Grade 3 TEAEs were decreased neutrophil count (18.6%) and anemia (13.3%). Among 89 evaluable Claudin18.2-positive patients (those who underwent ≥1 imaging assessment post-treatment), the ORR was 32.6% (Table 6). For all 93 Claudin18.2-positive patients, the median follow-up time was 5.98 months, with a median PFS of 4.76 months (95% CI 3.35-6.14). The median OS was not reached, and the 9-month OS rate was 56.4%.
Conclusion:In summary, CMG901 has demonstrated promising clinical efficacy in Claudin18.2-positive G/GEJ patients, with a controllable safety profile. These findings provide strong support for further evaluation of the efficacy and safety of CMG901 in Claudin18.2-positive G/GEJ patients.
Clinical trial information: NCT04805307
Table 6 Efficacy of Different Doses of CMG901 in Claudin18.2-Positive G/GEJ Patients
Abstract Number: TPS4182
GEMINI-Gastric: A Phase II Study of a Novel Combination Therapy for Patients with Unresectable Locally Advanced or Metastatic Gastric Cancer
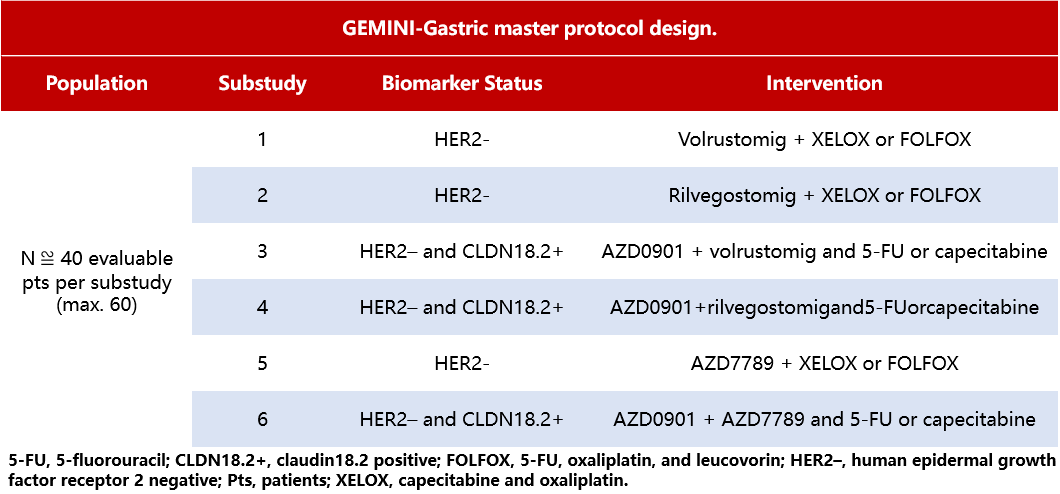
Background:Most gastric cancer cases are diagnosed at locally advanced stages or with distant metastases, resulting in a low survival rate. Currently, doublet chemotherapy (CTx) based on platinum and fluoropyrimidine remains the standard treatment for advanced gastric cancer. According to recent clinical data, IO combined with CTx can improve overall survival (OS) in patients with HER2-negative/low expression (HER2-) unresectable locally advanced or metastatic gastric cancer or gastroesophageal junction (GEJ) adenocarcinoma who have not previously received systemic therapy. GEMINI-Gastric (NCT05702229) is a Phase II, open-label, multi-drug, multi-center, master protocol study designed to evaluate the efficacy, safety, tolerability, pharmacokinetics (PK), and immunogenicity of various novel IO+CTx combination therapies.
Method:Select adult patients (≥18 years) with unresectable locally advanced or metastatic G/GEJ who have not previously received HER2-targeted therapy. According to RECIST v1.1 criteria, all enrolled patients should have measurable target lesions and an ECOG-PS score of 0-1. The design of this study allows for the evaluation of multiple novel drug combinations with CTx across different sub-studies (Table 7). Specific novel agents include: volrustomig, a bispecific humanized IgG1 monoclonal antibody (mAb) targeting PD-1/CTLA-4; rilvegostomig, a bispecific humanized IgG1 mAb targeting PD-1/TIGIT; AZD0901, a Claudin18.2-targeting ADC; and AZD7789, a bispecific mAb targeting PD-1/TIM-3. Per RECIST v1.1 criteria, the co-primary endpoints are investigator-assessed ORR at 6 months and PFS; secondary endpoints include safety, DOR, PFS, OS, PK, and immunogenicity. The sample size has been selected to provide sufficient precision for the primary endpoints. This study is currently recruiting at research institutions in the United States, Europe, and Asia.
Clinical Trial Information: NCT05702229
Table 7 GEMINI-Gastric Study Design
Abstract No.: TPS3163
CLARITY-PanTumor01 Study: Phase II Clinical Trial of Claudin18.2-Specific Antibody-Drug Conjugate AZD0901 (Formerly Known as CMG901) for the Treatment of Patients with Advanced Solid Tumors Expressing Claudin18.2
Background:Gastric/Gastroesophageal Junction (G/GEJ) cancer and pancreatic cancer have poor prognoses, with significant unmet clinical needs. Claudin18.2 (CLDN18.2) is highly expressed in gastric cancer, esophageal cancer, and pancreatic ductal adenocarcinoma (PDAC), and is a clinically validated target. AZD0901 is a potential first-in-class antibody-drug conjugate (ADC), consisting of a humanized anti-CLDN18.2 IgG1 antibody linked to the cytotoxic microtubule inhibitor MMAE payload via a protease-cleavable linker. Interim results from the first-in-human Phase I clinical study (NCT04805307) of single-agent AZD0901 in patients with G/GEJ cancer who are refractory and/or intolerant to standard treatments demonstrated promising efficacy and manageable safety. This study is an ongoing Phase II clinical trial evaluating AZD0901 for the treatment of patients with CLDN18.2-expressing advanced solid tumors, including G/GEJ cancer and PDAC.
Method:This study is an open-label, multi-center research comprising multiple sub-studies. Sub-study 1 is enrolling patients with HER2-negative and CLDN18.2-expressing G/GEJ cancer, including unresectable or metastatic patients who have received ≤2 prior lines of therapy. Patients are randomized 1:1 to receive AZD0901 at a dose of 1.8mg/kg or 2.2mg/kg via intravenous (IV) infusion once every three weeks (Q3W). Sub-study 2 is enrolling treatment-naïve metastatic PDAC patients expressing CLDN18.2, administering AZD0901 (maximum dose 2.2mg/kg, IV, Q3W), and combining it, based on investigator choice, with either 5-fluorouracil (2400mg/m², IV, days 1 and 2, Q2W), leucovorin (400mg/m²), or levoleucovorin (200mg/m²) (IV, days 1 and 2, Q2W), irinotecan (150 or 180mg/m²), or liposomal irinotecan (50mg/m²) (IV, day 1, Q2W) (Group 1), or gemcitabine (1000mg/m², IV, days 1 and 8, Q3W) (Group 2).
Treatment will continue until disease progression, intolerable toxicity occurs, or the patient withdraws informed consent. Eligible patients are aged ≥18 years with histologically confirmed unresectable or metastatic disease, having ≥1 measurable lesion according to RECIST v1.1 criteria, and an ECOG performance status of 0-1. Key exclusion criteria include unstable and/or active peptic ulcer disease or gastrointestinal bleeding, ascites requiring drainage, central nervous system metastases, and prior treatment with MMAE-based ADCs or CLDN18.2-targeted agents (excluding monoclonal antibodies targeting CLDN18.2). The primary endpoints include safety, tolerability, and ORR assessed per RECIST v1.1. Secondary endpoints include OS, PFS, duration of response, disease control rate, best percentage change in target lesion size, pharmacokinetics, immunogenicity, and pharmacodynamics. Recruitment began in December 2023, with patients being enrolled at study sites in Australia, Asia, Europe, and North America.
Clinical trial information: NCT06219941


 More medical information, click"Read the original text"View
More medical information, click"Read the original text"View