Novo Nordisk Breakthrough: Two Weekly Formulations Approved in China; CSPC, BeiGene, Hengrui, AstraZeneca... Racing for 2024 NRDL Negotiations | Insight Weekly Drug Report

Johnson & Johnson
Medical Device R&D and Manufacturer

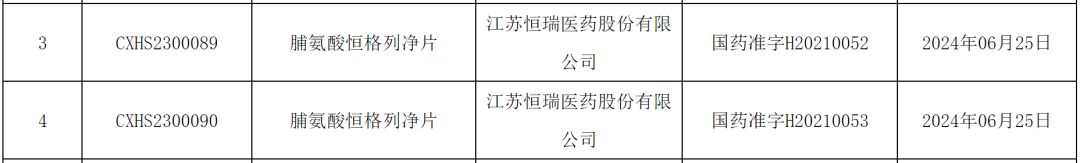
According to the Insight database statistics, this week (June 23 - June 29), a total of 48 innovative drugs (including improved new drugs) worldwide have advanced to new stages of development, 7 of which...One approved for marketing, three submitted for marketing approval, 178 products approved for clinical trials, 8 products applied for clinical trials.
The following text will introduce some key projects from both within and outside China.

Progress of Innovative Drugs in China
The adjustment work for the 2024 medical insurance catalog has already begun, and the deadline for innovative drugs to be eligible for access negotiations is June 30. Therefore, as expected, in the last week before the "deadline,"This week has also become a week of a surge in new drug approvals, with countless new drugs roaring across the finish line and successfully completing their final push.

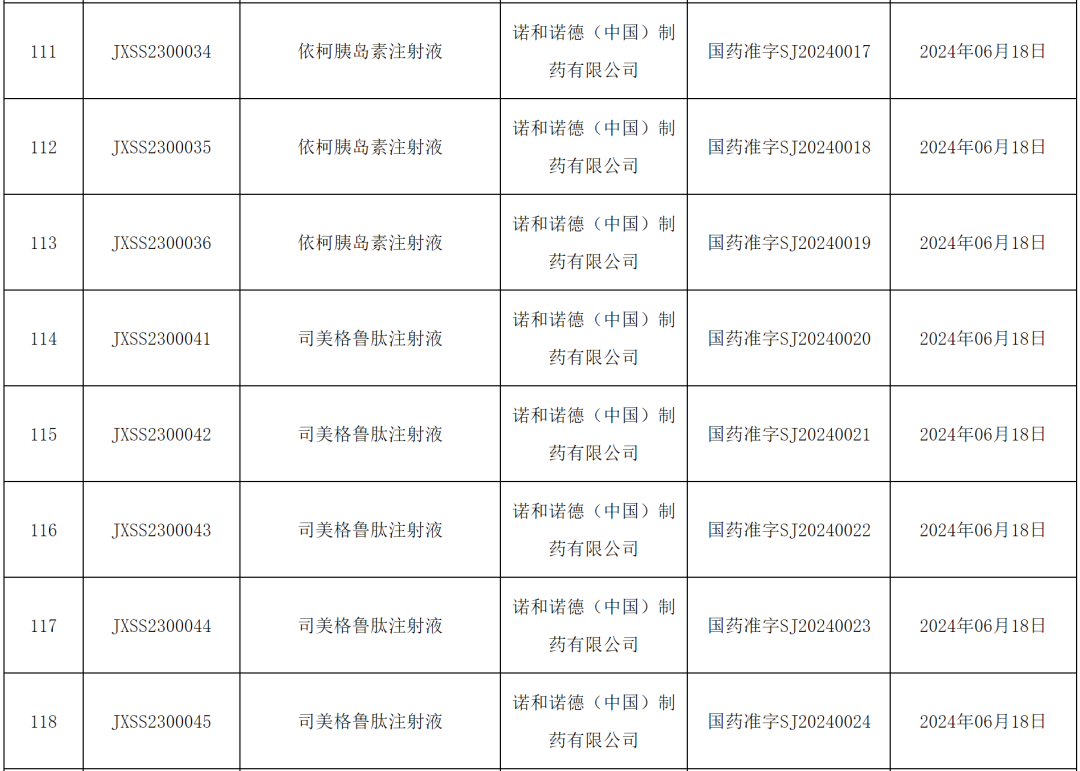
From the perspective of the first approval of a new drugJohnson & Johnson's fourth new multiple myeloma drug, the CD3/BCMA bispecific antibody "Talquetamab"; Biotech's Class 1 new drug "Bevibapeptide"; Haisco's DPP4 inhibitor "Cagliatin"; and CSPC's PD-1 monoclonal antibody "Envafolimab"… were all approved for marketing this week. Of course, Novo Nordisk's two weekly formulations "Icodec insulin" and the weight-loss version of "Semaglutide" are blockbuster-level drugs.
From the perspective of new indications approval,This week, Junshi's PD-1 monoclonal antibody, Beigene's PD-1 monoclonal antibody, Merck's PD-1 monoclonal antibody, AstraZeneca's third-generation EGFR-TKI, and Hengrui's SGLT2 inhibitor… all have new indications approved for marketing.
Therefore, this week's Insight New Drug Weekly Report will mainly introduce the approved information.
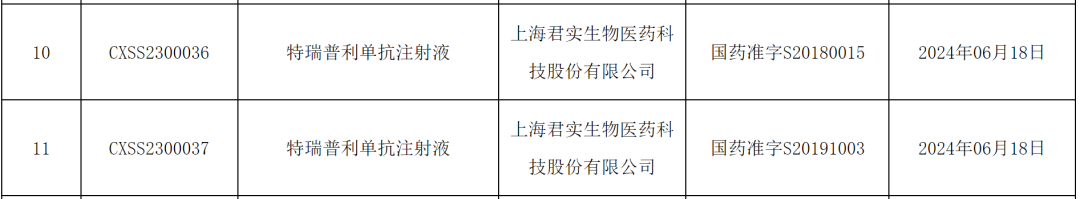
In addition, this week in China, there are also 15A new drug has been approved for clinical trials, and 7 drugs have applied for clinical trials.
15 Innovative Drugs (Including Modified New Ones) Granted Clinical Approval for the First Time in China This Week

Source: Insight Database Web Version
(The following text is from the same source unless otherwise specified.)
New Imported Drug Approved for the First Time
1. Johnson & Johnson: CD3/BCMA Bispecific Antibody Approved in China for the First Time
On June 25, Johnson & Johnson announced that its innovative therapeutic drug Tecvayli® (Teclistamab Injection) has been officially approved by the NMPA as a monotherapy for adult patients with relapsed or refractory multiple myeloma (RRMM) who have received at least three prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.

Screenshot source: Johnson & Johnson Official WeChat
Talike® is the first bispecific antibody approved for the treatment of RRMM, targeting B-cell maturation antigen (BCMA) and CD3, and has previously been approved for marketing in countries or regions such as the United States and the European Union.
This is a globally pioneering, ready-to-use, weight-based subcutaneous injection bispecific antibody that redirects CD3+ T cells to BCMA-expressing myeloma cells to induce tumor cell killing. Clinically validated, Tecvayli® demonstrates a high response rate in patients with relapsed or refractory multiple myeloma who have received at least three prior lines of therapy, with even greater benefits for Chinese patients, achieving an overall response rate (ORR) of 76.9%.
Talike® is the fourth product approved in China by Johnson & Johnson for the treatment of multiple myeloma., further enriching its leading product portfolio in the field of multiple myeloma and hematological malignancy treatment, allowing more patients to have the opportunity to achieve functional cure.

New Drug Made in China Approved for Marketing for the First Time
1、CSPC: PD-1 Monoclonal Antibody Approved for Marketing
On June 28, the NMPA website showed that the Enlansubai monoclonal antibody injection developed by CSPC.(Product Name: Enshuxing)Approved for marketing to treat patients with PD-L1 positive recurrent or metastatic cervical cancer who have failed at least one line of platinum-based chemotherapy.


 Screenshot from: NMPA official website
Screenshot from: NMPA official website
Enlangsubai monoclonal antibody is a fully human, high-affinity anti-PD-1 IgG4 monoclonal antibody, which has been confirmed through early-phase Ib studies to exhibit favorable efficacy in PD-L1 positive recurrent or metastatic cervical cancer patients. The phase II study presented at the ASCO 2024 conference.(NCT04886700)Further verifying its safety and efficacy.
This Phase II study enrolled a total of 107 patients who were PD-L1 positive(CPS≥1)Patients with cervical cancer who experienced disease progression or intolerance to first-line platinum-based therapy during or after receiving it. Patients received 240 mg of enfortumab vedotin every two weeks, for a maximum treatment duration of 24 months, until disease progression, intolerable toxicity, or other study termination criteria were met.
The results showed that, after a median follow-up of 13.96 months, the ORR and DCR of the patients were 29.0% (95% CI, 20.6-38.5) and 54.2% (95% CI, 44.3-63.9), respectively, including two complete responses and 29 partial responses. The mDoR was 16.6 months (95% CI, 10.8-NA), and the mPFS was 3.1 months (95% CI, 2.2-6.9).
In terms of safety, 97.2% of patients experienced at least one treatment-related adverse event, and immune-related adverse events were observed in 41.1% of patients.(irAEs), No treatment-related deaths occurred.
 Screenshot source: Insight Database official website
Screenshot source: Insight Database official website
It is reported that Enlangsubai monoclonal antibody combined with platinum-based chemotherapy ± Bevacizumab as first-line treatment for PD-L1 positive.(CPS≥1)Phase III Clinical Study of Recurrent or Metastatic Cervical Cancer(CTR20230132/NCT05715840)Launched in January 2023. This study aims to evaluate the safety, tolerability, and efficacy of the treatment regimen in such patients, with the hope of providing new therapeutic options and clinical guidance for this group.
2. Bio-Thera: "Bevibaptide" Approved for Marketing
On June 28, Bio-Thera announcedReceived approval issued by NMPA regardingBivalirudin Citrate Injection (BAT2094, Trade Name: Betanin®, Formerly Known as Batifiban)《Drug Registration Certificate》, for patients with acute coronary syndrome undergoing percutaneous coronary intervention (including coronary stent placement), to reduce the risk of acute occlusion, stent thrombosis, no-reflow, and slow-flow.
This drug is a Class 1 chemical medicine independently developed by Bio-Thera with proprietary intellectual property rights. It is a peptide-based β3 integrin receptor inhibitor.


Screenshot from: NMPA official website
Glycoprotein receptor αIIbβ3 (also known as IIb/IIIa) is a platelet surface receptor that plays a dominant role in the process of platelet aggregation. Citrate bevibaptide prevents fibrinogen, Von Willebrand factor, and other adhesive ligands from binding to glycoprotein receptor αIIbβ3, thereby blocking platelet cross-linking and platelet aggregation. At the same time, citrate bevibaptide can also inhibit integrin receptor αvβ3, which is associated with vascular wall cell proliferation, thus suppressing the growth of vascular smooth muscle and reducing the risk of arterial re-occlusion.
The efficacy and safety of Citric Acid Bevibaptide Injection have been demonstrated through a multicenter, randomized, double-blind, placebo-controlled parallel design study.Phase II Clinical Trial, and a multi-center, randomized, double-blind, placebo-controlled parallel designPhase III Clinical TrialConfirm.
Results from the Phase II clinical study showed that, in PCI patients, a single intravenous administration of 220 µg/kg and a continuous intravenous infusion at 2.5 µg/min/kg of this product during the perioperative period led to measurements of platelet aggregation inhibition rates at 4 h and 24 h post-surgery. It was found that, compared with the placebo group, the administration of Bivalirudin Citrate rapidly increased the platelet aggregation inhibition rate to over 80%. This rate remained stable during the constant-rate infusion and quickly reverted to normal platelet aggregation function after the infusion ended.
The primary efficacy endpoint of the Phase III clinical trial for Citrate Bevilaptide Injection was a composite endpoint including death, myocardial infarction, acute target vessel revascularization, thrombotic treatment requirements due to complications such as acute occlusion, no-reflow, or similar PCI-related issues, and no-reflow or severe slow-flow within 30 days post-PCI. The HR for the composite endpoint events in the Citrate Bevilaptide group compared to the control group was 0.57 (95% CI: 0.35, 0.94, P=0.026), indicating that Citrate Bevilaptide Injection reduced the risk of composite endpoint events by more than 43% on top of existing standard treatments.
3、Haisco: DPP4 Inhibitor "Cagrilintide" Approved for Marketing
On June 24, according to the NMPA official website, the 1st-class innovative drug Cogrelletin tablets submitted by Haisco Pharmaceutical were approved.(Product Name: Beichangping)Launched to improve blood glucose control in adult patients with type 2 diabetes.
 Screenshot from: NMPA official website
Screenshot from: NMPA official website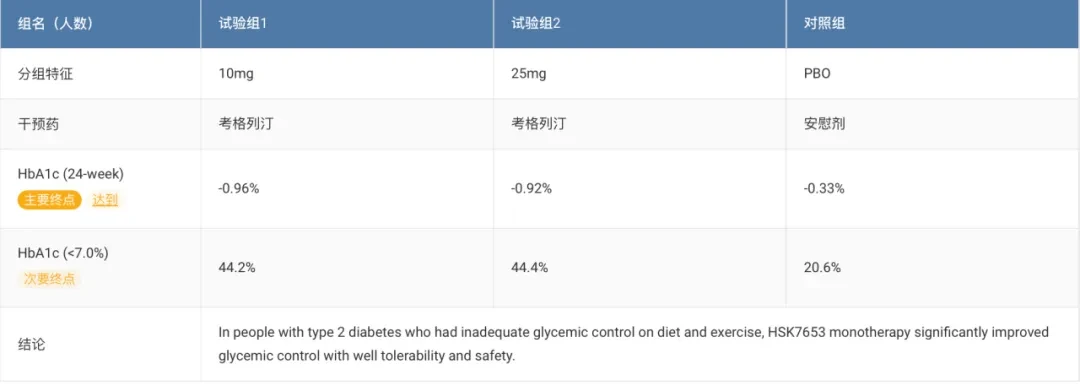 Screenshot source: Insight Database official website
Screenshot source: Insight Database official websiteNew Indication Approved for Marketing
1、AstraZeneca: "Osimertinib" Receives Another New Indication Approval! First-Line Combination with Chemotherapy for EGFR-Mutated NSCLC
On June 26, AstraZeneca announced that the NMPA has officially approved TAGRISSO® (generic name: Osimertinib Mesylate Tablets) in combination with pemetrexed and platinum-based chemotherapy for the first-line treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) harboring epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations.


This is the fourth indication for osimertinib approved in China. The drug was previously approved for: 1) Second-line treatment of locally advanced or metastatic NSCLC with disease progression during or after prior EGFR-TKI therapy and the presence of EGFR T790M mutation; 2) First-line treatment of patients with locally advanced or metastatic NSCLC harboring EGFR exon 19 deletion or exon 21 (L858R) substitution mutations; 3) Adjuvant treatment (with or without adjuvant chemotherapy as determined by the physician) for patients with stage IB-IIIA NSCLC with EGFR exon 19 deletion or exon 21 (L858R) substitution mutations following complete tumor resection. Furthermore, all three indications have been included in the National Reimbursement Drug List.
This new indication approval is mainly based on the results of the global multicenter Phase III randomized controlled study FLAURA2 published in NEJM in 2023, the efficacy and safety results of the China cohort consistent with the global cohort results announced at the 2023 European Society for Medical Oncology Asia Congress (ESMO-ASIA), and the analysis results of the central nervous system (CNS) efficacy of the FLAURA2 study published in the Journal of Clinical Oncology.
In the FLAURA2 full-team cohort (N=557), results assessed by investigators showed that, compared with osimertinib monotherapy, the combination of osimertinib and chemotherapy reduced the risk of disease progression or death by 38%. The median progression-free survival (mPFS) was extended by 8.8 months in the osimertinib plus chemotherapy group compared to the osimertinib monotherapy group (25.5 months vs. 16.7 months) (HR 0.62; 95% CI 0.49-0.79; p<0.0001). Results assessed by the Blinded Independent Central Review (BICR) were consistent with those assessed by investigators: mPFS was extended by 9.5 months in the osimertinib plus chemotherapy group compared to the osimertinib monotherapy group (29.4 months vs. 19.9 months) (HR 0.62; 95% CI 0.48-0.80; p=0.0002).
Although the OS results are not yet mature, the interim analysis of OS updated at the 2024 European Lung Cancer Conference (ELCC) (with 41% maturity) showed an encouraging trend in OS benefit (HR 0.75; 95% CI 0.57-0.97). In terms of safety, the overall safety profile of first-line treatment with osimertinib combined with chemotherapy was good. Adverse events (AEs) were mainly related to chemotherapy, and the proportion of patients discontinuing osimertinib due to adverse events was low in both groups: 11% in the osimertinib plus chemotherapy group and 6% in the osimertinib monotherapy group.
Efficacy and Safety of the FLAURA2 Study in the Chinese Cohort Show Consistent Results with the Global Cohort. Notably, in the Chinese cohort (N=131), results assessed by investigators showed that the risk of disease progression or death was reduced by 44% in the osimertinib plus chemotherapy group compared to the osimertinib monotherapy group (HR 0.56; 95% CI 0.34-0.92). The results assessed by BICR were generally consistent with those evaluated by investigators. According to BICR assessment, the osimertinib plus chemotherapy group reduced the risk of disease progression or death by 42% compared to the osimertinib monotherapy group (HR 0.58; 95% CI 0.34-1.01). In terms of safety, the overall profile was consistent with the global cohort.
As of now, the indication for osimertinib combined with chemotherapy as a first-line treatment for patients with EGFR-mutated advanced NSCLC was approved by the FDA on February 16, 2024, and has been unanimously recommended by the National Comprehensive Cancer Network (NCCN) Guidelines and the Chinese Society of Clinical Oncology (CSCO) Guidelines.

The approval of the obinutuzumab SDI dosing regimen is based on the international multicenter Phase IV clinical GAZELLE study. Data show that the safety of the obinutuzumab SDI regimen is favorable: no patients experienced IRRs of Grade 3 or higher during Cycle 2 of SDI, and only one patient experienced Grade 3 IRR in subsequent treatment cycles, with no patients experiencing Grade 4 or 5 IRR.
In terms of time, the median infusion duration for the SDI regimen is 95-98 minutes, which is more than half shorter compared to the standard infusion regimen of 205-269.5 minutes. In terms of efficacy, the ORR for the SDI regimen is 86.7%, consistent with the data reported in the Phase III GALLIUM study (88.5%) Consistent.
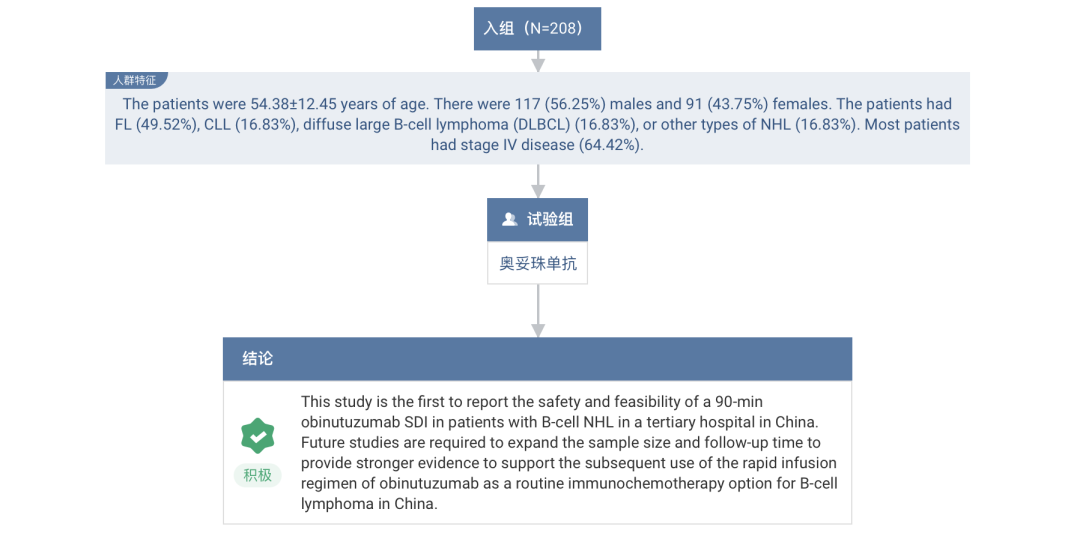
Another study conducted in hospitals in China enrolled 208 patients with advanced non-Hodgkin lymphoma (NHL) patients. The results showed that Obinutuzumab 90-minute SDI was well tolerated. Among 814 infusions, only 2 cases (0.25%) IRR. This study further confirms the safety of the Obinutuzumab SDI regimen in Chinese patients.
 Screenshot source: Insight Database official website
Screenshot source: Insight Database official website
Overall, the successful approval of the Obinutuzumab SDI regimen in China not only provides patients with a more convenient and efficient treatment option, but also further reduces the burden of the disease on patients, while enhancing the clinical management level of hematological tumors in China.
3, BeiGene: The 13th Indication for PD-1 Monoclonal Antibody Approved for Marketing
On June 28, BeiGene announced that its PD-1 inhibitor, BeiGene's Baizean® (tislelizumab), has officially received NMPA approval for use in combination with etoposide and platinum-based chemotherapy as a first-line treatment for extensive-stage small cell lung cancer (ES-SCLC).
This is the 14th approved indication for Tislelizumab, with 11 of them already included in the National Medical Insurance Drug List, making it the PD-1 inhibitor with the highest number of approved indications included in the National Medical Insurance Drug List.



This approval is based on the clinical trial data from RATIONALE 312 (NCT04005716). RATIONALE 312 is a randomized, double-blind, placebo-controlled, multi-center Phase 3 clinical trial comparing the efficacy of tislelizumab combined with chemotherapy versus placebo combined with chemotherapy as a first-line treatment for patients with extensive-stage small cell lung cancer. The trial enrolled 457 patients across 51 centers in China.
The study results showed that: Compared with chemotherapy, tislelizumab combined with chemotherapy significantly improved the overall survival (OS) of patients with ES-SCLC. The median OS for the tislelizumab plus chemotherapy group was 15.5 months (13.5 months in the chemotherapy group, HR: 0.75), with a 3-year OS rate reaching 25% (chemotherapy group: 9%), and demonstrated a favorable safety profile. The results of this study were presented as a late-breaking oral report at the World Conference on Lung Cancer (WCLC) held in Singapore in 2023 and were recently published in the prestigious journal *Journal of Thoracic Oncology* in the field of lung cancer.
Outside of China, tislelizumab has been approved in the European Union, the United Kingdom, the United States, South Korea, Switzerland, and Australia. The supplemental New Drug Application for its first-line treatment of gastric or gastroesophageal junction cancer and the supplemental New Drug Application for its first-line treatment of esophageal squamous cell carcinoma are currently under review by the U.S. Food and Drug Administration (FDA) and the European Commission (EC).
4. Junshi Biosciences: The 10th Indication of PD-1 Monoclonal Antibody Approved for Marketing
On June 25, Junshi Biosciences announced that the new indication for first-line treatment of recurrent or metastatic triple-negative breast cancer (TNBC) with PD-L1 positivity (CPS≥1) as assessed by a fully validated test has been approved by the NMPA. This involves the combination of the company's self-developed anti-PD-1 monoclonal antibody drug, Toripalimab Injection (Tuoyi®), with Albumin-bound Paclitaxel for Injection.
This is the 10th indication approved for Toripalimab in China.


The approval of this new indication is mainly based on the data results of the TORCHLIGHT study (NCT04085276). The TORCHLIGHT study is a randomized, double-blind, placebo-controlled, multi-center Phase III clinical study, led by Professor Jiang Zefei, Vice Chairman and Secretary General of the Chinese Society of Clinical Oncology (CSCO) and Director of the Department of Oncology at the PLA General Hospital, and conducted across 56 centers in China.
In February 2023, the Independent Data Monitoring Committee (IDMC) determined that the primary endpoint of the TORCHLIGHT study reached the pre-specified superiority boundary at the interim analysis, making it the first Phase III registrational study in China to yield positive results in the field of advanced TNBC immunotherapy.
In January 2024, the international top-tier medical journal *Nature Medicine* (Impact Factor: 58.7) published the interim analysis data of the TORCHLIGHT study. The results showed that compared with injectable paclitaxel (albumin-bound), toripalimab combined with injectable paclitaxel (albumin-bound) significantly prolonged the progression-free survival (PFS) in PD-L1 positive patients with first-diagnosed stage IV or recurrent metastatic TNBC. Overall survival (OS) also showed a trend of benefit, achieving a breakthrough in the immunotherapy of advanced TNBC in China.
Among them, the median PFS in the toripalimab group reached 8.4 months, with a 35% reduction in the risk of disease progression or death (P=0.0102); the median OS in the toripalimab group was extended by 13.3 months (32.8 months vs 19.5 months), with a 38% reduction in the risk of death (P=0.0148). The safety data of toripalimab was consistent with the known risks, and no new safety signals were identified.
5. Hengrui Medicine: "Hengge Liejing" New Indication Approved for Marketing
On June 25, the NMPA website announced that Hengrui Medicine's "Hengli Gliflozin" has been approved for a new indication, in combination with Rimegliflozin Phosphate and Metformin, for adult patients with type 2 diabetes whose blood sugar levels remain uncontrolled despite treatment with Metformin alone. The marketing application for this indication was initially accepted on September 27, 2023.



On June 24, Haihe Pharmaceuticals announced that the company's innovative drug MET inhibitor Glumetinib has been approved for marketing by Japan's Ministry of Health, Labour and Welfare for the treatment of locally advanced or metastatic non-small cell lung cancer with MET exon 14 skipping mutations.
The drug was discovered by the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences, independently developed by Shanghai Haihe Biopharma Co., Ltd., which owns global independent intellectual property rights.

The marketing authorization application for this indication is primarily based on the efficacy and safety data from the pivotal Phase II SCC244-108 study (GLORY study, NCT04270591). This study is a multinational, multicenter, open-label, single-arm clinical trial designed to evaluate the efficacy and safety of glutitinib tablets in patients with locally advanced or metastatic non-small cell lung cancer harboring MET exon 14 skipping mutations. This indication was approved for marketing by China's National Medical Products Administration (NMPA) on March 7, 2023. The global principal investigator of this study is Professor Lu Shun from the Shanghai Chest Hospital, affiliated with the Shanghai Jiao Tong University School of Medicine.
As of April 28, 2022, the 12-month follow-up data from 79 patients with METex14 skipping mutations confirmed by the central laboratory in the GLORY study showed: The overall objective response rate (ORR) assessed by the Blinded Independent Review Committee (BIRC) was 65.8%, with an ORR of 70.5% in treatment-naïve patients and 60.0% in previously treated patients; the median progression-free survival (mPFS) in the overall population was 8.5 months, reaching 11.7 months in treatment-naïve patients and 7.6 months in previously treated patients; the median overall survival (mOS) in the overall population was 17.3 months, not yet reached in treatment-naïve patients and 16.2 months in previously treated patients, demonstrating clear efficacy.
In terms of safety, the overall safety is well-tolerated. Common adverse reactions include edema, headache, gastrointestinal symptoms, and elevated liver transaminases, mostly mild to moderate, which can be alleviated or resolved after symptomatic treatment. There is no potential phototoxicity, nor have related allergic reactions been observed. The drug has minimal interactions and a lower risk of safety issues when used in combination with other medications.

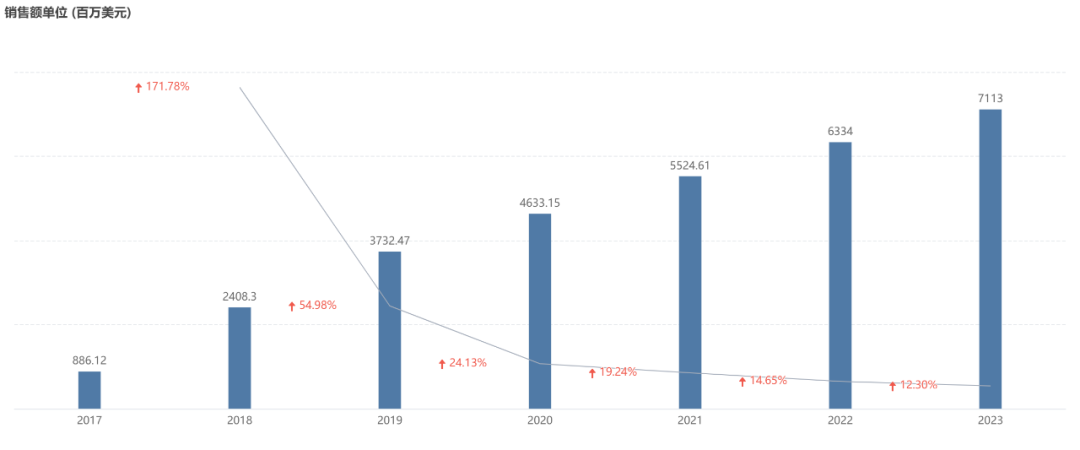
Ocrevus Global Sales in Recent Years