According to statistics from the Insight database, this week (September 15th - September 21st), a total of 72 innovative drugs (including improved new drugs) worldwide have advanced to new stages of development, with 3One product has been submitted for marketing approval, three products have entered Phase III clinical trials, and sixteenFive products approved for clinical trials, five products submitted for clinical trials.
The following text will introduce some key projects from both within and outside China.

Progress of Innovative Drugs Overseas
Outside of China, several new drugs/new indications have been newly approved this week. ESMO clinical results are still being released continuously—see Insight's recent reports for details. Meanwhile, this weekly report will focus on important R&D progress and clinical results outside of ESMO that should not be missed, providing readers with sharing and supplementary information.。
Approved for Marketing
According to the Insight database, this week Johnson & Johnson's重磅双抗新药埃万妥单抗(Amivantamab)has received another approval in the United States for a new indication, used as a second-line treatment for NSCLC. 帕博利珠单抗 (K药) has expanded its indications again, now for first-line treatment of pleural mesothelioma. Novartis' CDK4/6 inhibitor 瑞波西利 has received regulatory approval for a significant adjuvant treatment indication, which will help them continue to make advances in the CDK field.
In addition, AstraZeneca's IL-5R monoclonal antibody Benralizumab has been newly approved for an indication, and Bavarian Nordic also has a vaccine approved in Europe. Details are as follows:
Screenshot source: Insight Database Web Version1. Novartis: CDK4/6 Inhibitor "Ribociclib" Approved for New Indication, Adjuvant Treatment for Breast CancerOn September 17, Novartis announced that the new indication for CDK4/6 inhibitor Ribociclib (Kisqali) has been approved by the FDA for adjuvant treatment of early HR+/HER2- breast cancer.Screenshot source: Official website of the companyRibociclib (also known as LEE011)It is the second CDK4/6 inhibitor approved for marketing globally. The first one is Pfizer's Palbociclib, which reached a peak sales of $5.437 billion in 2021, capturing 70% of the market share for this target. The closely following Ribociclib and Abemaciclib are also formidable, experiencing rapid growth and continuously eroding its market share. Eli Lilly's Abemaciclib, in particular, has shown astonishing growth, with sales reaching $3.863 billion in the 2023 fiscal year, and itsOne of the key factors for rapid growth is precisely due to the early breakthrough in adjuvant treatment indications.This time, Novartis has finally been approved for adjuvant treatment, and its growth potential is self-evident.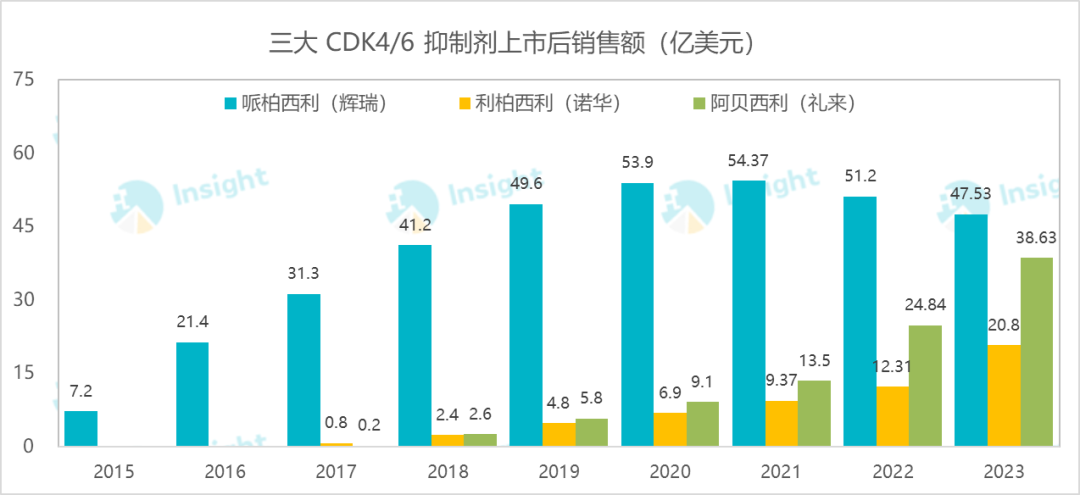 Novartis previously disclosed the Phase III trial of ribociclib combined with non-steroidal aromatase inhibitors for adjuvant treatment in HR+/HER2- early breast cancer patients at the 2023 ASCO.NATALEE StudyResults. The data shows that Ribociclib + Non-steroidal aromatase inhibitor (NSAI) treatment shows a significant advantage over NSAI alone in terms of invasive disease-free survival (iDFS) (HR=0.748;95% CI,0.618-0.906;P =0.0014)。At the 2023 SABCS meeting, Novartis disclosed the latest iDFS data.As of July 21, 2023, among the 2,549 patients in the ribociclib (RIB) + nonsteroidal aromatase inhibitor (NSAI) group, 2.8% completed 3 years of RIB treatment, 35.5% discontinued RIB or RIB+NSAI early, and 20.7% were still using RIB.NSAI Group 68.5% Continued Treatment.The results showed that, with a median follow-up of 33.3 months, RIB+NSAI significantly reduced the risk of disease recurrence by 25.1% compared to NSAI alone (HR=0.749; 95% CI: 0.628-0.892; p=0.0006). The 3-year iDFS rates were 90.7% in the RIB+NSAI group and 87.6% in the NSAI group. Consistent benefits were also observed across all secondary efficacy endpoints, including a 25.1% reduction in the risk of distant disease-free survival (DDFS) and a 27.3% reduction in the risk of recurrence-free survival (RFS).In terms of safety, no new safety signals were observed.2. Johnson & Johnson: Amivantamab Receives Approval for Another New Indication9 On the 19th, Johnson & Johnson announced that the FDA had approved its EGFR/c-Met bispecific antibody Amivantamab (RYBREVANT®) in combination with standard chemotherapy for the treatment of adult patients with locally advanced or metastatic NSCLC who have EGFR exon 19 deletions or L858R substitution mutations and are progressing on or after EGFR-TKI therapy.This combination previously gained widespread attention for its head-to-head "sniping" of AstraZeneca's blockbuster drug, osimertinib. On August 20 this year, the drug was just approved by the FDA for use in combination with third-generation EGFR-TKI as a first-line treatment for EGFRm NSCLC. The current approval is for later-line treatment with osimertinib, making its coverage of this type of lung cancer more comprehensive.Screenshot from: Johnson & Johnson official websiteThe approval of this new indication is based on MARIPOSA-2Study (NCT04988295), which is an Amivantamab (Rybrevant/Amivantamab) A Phase III trial of combination chemotherapy for NSCLC patients with resistance to osimertinib, which achieved dual primary endpoints. This is the first Phase III study to demonstrate clinically meaningful PFS improvement in post-osimertinib treatment. The results showed that, compared with chemotherapy alone, the combination therapy reduced the risk of disease progression or death by 52%, which was the primary endpoint of the trial. The PFS was 6.3 months vs. 4.2 months. The ORR in the combination therapy group was 53%, while it was only 29% in the chemotherapy-alone group.According to the Insight database, Amivantamab has been successively approved for multiple indications in Europe and the US. Just this year alone, this is already the third indication approved by the FDA, which can be said to comprehensively cover the EGFRm NSCLC field. In China, Johnson & Johnson has also submitted3 marketing applications.
Novartis previously disclosed the Phase III trial of ribociclib combined with non-steroidal aromatase inhibitors for adjuvant treatment in HR+/HER2- early breast cancer patients at the 2023 ASCO.NATALEE StudyResults. The data shows that Ribociclib + Non-steroidal aromatase inhibitor (NSAI) treatment shows a significant advantage over NSAI alone in terms of invasive disease-free survival (iDFS) (HR=0.748;95% CI,0.618-0.906;P =0.0014)。At the 2023 SABCS meeting, Novartis disclosed the latest iDFS data.As of July 21, 2023, among the 2,549 patients in the ribociclib (RIB) + nonsteroidal aromatase inhibitor (NSAI) group, 2.8% completed 3 years of RIB treatment, 35.5% discontinued RIB or RIB+NSAI early, and 20.7% were still using RIB.NSAI Group 68.5% Continued Treatment.The results showed that, with a median follow-up of 33.3 months, RIB+NSAI significantly reduced the risk of disease recurrence by 25.1% compared to NSAI alone (HR=0.749; 95% CI: 0.628-0.892; p=0.0006). The 3-year iDFS rates were 90.7% in the RIB+NSAI group and 87.6% in the NSAI group. Consistent benefits were also observed across all secondary efficacy endpoints, including a 25.1% reduction in the risk of distant disease-free survival (DDFS) and a 27.3% reduction in the risk of recurrence-free survival (RFS).In terms of safety, no new safety signals were observed.2. Johnson & Johnson: Amivantamab Receives Approval for Another New Indication9 On the 19th, Johnson & Johnson announced that the FDA had approved its EGFR/c-Met bispecific antibody Amivantamab (RYBREVANT®) in combination with standard chemotherapy for the treatment of adult patients with locally advanced or metastatic NSCLC who have EGFR exon 19 deletions or L858R substitution mutations and are progressing on or after EGFR-TKI therapy.This combination previously gained widespread attention for its head-to-head "sniping" of AstraZeneca's blockbuster drug, osimertinib. On August 20 this year, the drug was just approved by the FDA for use in combination with third-generation EGFR-TKI as a first-line treatment for EGFRm NSCLC. The current approval is for later-line treatment with osimertinib, making its coverage of this type of lung cancer more comprehensive.Screenshot from: Johnson & Johnson official websiteThe approval of this new indication is based on MARIPOSA-2Study (NCT04988295), which is an Amivantamab (Rybrevant/Amivantamab) A Phase III trial of combination chemotherapy for NSCLC patients with resistance to osimertinib, which achieved dual primary endpoints. This is the first Phase III study to demonstrate clinically meaningful PFS improvement in post-osimertinib treatment. The results showed that, compared with chemotherapy alone, the combination therapy reduced the risk of disease progression or death by 52%, which was the primary endpoint of the trial. The PFS was 6.3 months vs. 4.2 months. The ORR in the combination therapy group was 53%, while it was only 29% in the chemotherapy-alone group.According to the Insight database, Amivantamab has been successively approved for multiple indications in Europe and the US. Just this year alone, this is already the third indication approved by the FDA, which can be said to comprehensively cover the EGFRm NSCLC field. In China, Johnson & Johnson has also submitted3 marketing applications.
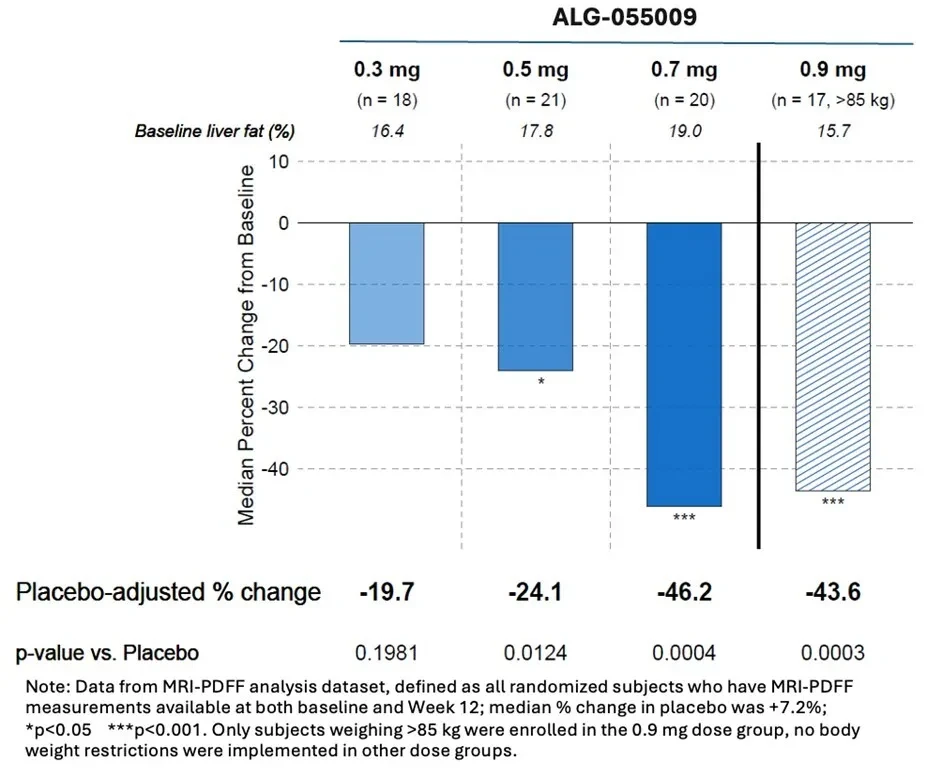
 1. Henlius: PD-1 Serplulimab Receives Positive CHMP Opinion in the EU, Approval Expected SoonOn September 20, Henlius announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has issued a positive opinion recommending the marketing authorization of the company's self-developed anti-PD-1 monoclonal antibody H drug Hansizhuang® (Serioli monoclonal antibody), suggesting its approval for first-line treatment of extensive-stage small cell lung cancer (ES-SCLC).In December 2022, H drug received orphan drug designation from the European Commission (EC) for the treatment of SCLC, and in March 2023, the EMA accepted the Marketing Authorization Application (MAA) for H drug for the treatment of SCLC in the EU.In 2023, Henlius reached a collaboration with Intas, granting it exclusive development and commercialization rights for H drug in over 50 countries across Europe and India. The recent positive opinion from the CHMP marks a significant step closer to achieving broader accessibility of H drug in Europe.The positive opinion for H drug's EU marketing authorization is mainly based on the ASTRUM-005 study. The ASTRUM-005 study is a randomized, double-blind, placebo-controlled international multicenter Phase III trial designed to evaluate the efficacy and safety of serplulimab combined with chemotherapy compared to placebo plus chemotherapy as a first-line treatment for ES-SCLC. This trial was conducted across 128 trial centers in multiple countries, including China, Poland in the EU, Turkey, and Georgia, enrolling 585 participants, of which approximately 31.5% were Caucasian.Results of the ASTRUM-005 clinical trial were first presented as an oral report at the 2022 American Society of Clinical Oncology (ASCO) Annual Meeting and published in The Journal of the American Medical Association (JAMA), one of the world's top four medical journals, marking it as the first SCLC immunotherapy clinical study to be featured in the main issue of JAMA. Based on the ASTRUM-005 study, Drug H has been approved in China and multiple Southeast Asian countries for the first-line treatment of ES-SCLC, becoming the world’s first anti-PD-1 monoclonal antibody approved for first-line treatment of small cell lung cancer.The globalization pace of Drug H is continuously accelerating. To date, the product has been approved for marketing in China, Indonesia, Cambodia, Thailand, and other countries. Its outward licensing covers more than 70 countries and regions, including the United States, Europe, Southeast Asia, the Middle East, and North Africa.As the first self-developed innovative monoclonal antibody by Henlius, H drug focuses on high-incidence cancer types such as lung cancer and gastrointestinal tumors. It has been approved in China for the treatment of microsatellite instability-high (MSI-H) solid tumors, squamous non-small cell lung cancer (sqNSCLC), extensive-stage small cell lung cancer (ES-SCLC), and esophageal squamous cell carcinoma (ESCC). Henlius is simultaneously conducting more than 10 clinical studies worldwide on immunotherapy combination regimens centered around H drug. Additionally, the company is carrying out a head-to-head bridging trial in the United States comparing H drug with the first-line standard treatment atezolizumab for ES-SCLC to further support the regulatory submission of H drug in the U.S. Heavyweight Clinical Results1. Liver fat reduced by up to 46.2%, positive results from Phase IIa trial of new MASH drugOn September 19, Aligos announced the positive topline results of its Phase IIa HERALD study of ALG-055009, a thyroid hormone receptor β (THR-β) agonist, in patients with Metabolic Dysfunction-Associated Steatohepatitis (MASH).In the 12th week, the median relative reduction in liver fat was as high as 46.2%, with a clear dose-response relationship.。Image Source:Corporate Website
1. Henlius: PD-1 Serplulimab Receives Positive CHMP Opinion in the EU, Approval Expected SoonOn September 20, Henlius announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has issued a positive opinion recommending the marketing authorization of the company's self-developed anti-PD-1 monoclonal antibody H drug Hansizhuang® (Serioli monoclonal antibody), suggesting its approval for first-line treatment of extensive-stage small cell lung cancer (ES-SCLC).In December 2022, H drug received orphan drug designation from the European Commission (EC) for the treatment of SCLC, and in March 2023, the EMA accepted the Marketing Authorization Application (MAA) for H drug for the treatment of SCLC in the EU.In 2023, Henlius reached a collaboration with Intas, granting it exclusive development and commercialization rights for H drug in over 50 countries across Europe and India. The recent positive opinion from the CHMP marks a significant step closer to achieving broader accessibility of H drug in Europe.The positive opinion for H drug's EU marketing authorization is mainly based on the ASTRUM-005 study. The ASTRUM-005 study is a randomized, double-blind, placebo-controlled international multicenter Phase III trial designed to evaluate the efficacy and safety of serplulimab combined with chemotherapy compared to placebo plus chemotherapy as a first-line treatment for ES-SCLC. This trial was conducted across 128 trial centers in multiple countries, including China, Poland in the EU, Turkey, and Georgia, enrolling 585 participants, of which approximately 31.5% were Caucasian.Results of the ASTRUM-005 clinical trial were first presented as an oral report at the 2022 American Society of Clinical Oncology (ASCO) Annual Meeting and published in The Journal of the American Medical Association (JAMA), one of the world's top four medical journals, marking it as the first SCLC immunotherapy clinical study to be featured in the main issue of JAMA. Based on the ASTRUM-005 study, Drug H has been approved in China and multiple Southeast Asian countries for the first-line treatment of ES-SCLC, becoming the world’s first anti-PD-1 monoclonal antibody approved for first-line treatment of small cell lung cancer.The globalization pace of Drug H is continuously accelerating. To date, the product has been approved for marketing in China, Indonesia, Cambodia, Thailand, and other countries. Its outward licensing covers more than 70 countries and regions, including the United States, Europe, Southeast Asia, the Middle East, and North Africa.As the first self-developed innovative monoclonal antibody by Henlius, H drug focuses on high-incidence cancer types such as lung cancer and gastrointestinal tumors. It has been approved in China for the treatment of microsatellite instability-high (MSI-H) solid tumors, squamous non-small cell lung cancer (sqNSCLC), extensive-stage small cell lung cancer (ES-SCLC), and esophageal squamous cell carcinoma (ESCC). Henlius is simultaneously conducting more than 10 clinical studies worldwide on immunotherapy combination regimens centered around H drug. Additionally, the company is carrying out a head-to-head bridging trial in the United States comparing H drug with the first-line standard treatment atezolizumab for ES-SCLC to further support the regulatory submission of H drug in the U.S. Heavyweight Clinical Results1. Liver fat reduced by up to 46.2%, positive results from Phase IIa trial of new MASH drugOn September 19, Aligos announced the positive topline results of its Phase IIa HERALD study of ALG-055009, a thyroid hormone receptor β (THR-β) agonist, in patients with Metabolic Dysfunction-Associated Steatohepatitis (MASH).In the 12th week, the median relative reduction in liver fat was as high as 46.2%, with a clear dose-response relationship.。Image Source:Corporate Website
HERALD (NCT06342947) is a randomized, double-blind, placebo-controlled trial that enrolled 102 subjects presumed to have MASH and stage 1-3 liver fibrosis. Subjects were randomized to receiveALG-055009 One of four doses (0.3, 0.5, 0.7, 0.9 mg) or a placebo, administered orally once daily for 12 weeks. The 0.9 mg dose group only recruited subjects weighing >85 kg, while there were no weight restrictions for the other dose groups.The primary endpoint was the relative change in liver fat content measured by MRI-PDFF at week 12. Safety, pharmacokinetics (PK), and other non-invasive biomarkers/tests previously shown to be affected by thyroid hormone receptor β (THR-β) agonist treatment were also evaluated.Doses of 0.5 mg to 0.9 mg of ALG-055009 showed a significant reduction in liver fat at week 12.After placebo adjustment, the median relative reduction measured by MRI-PDFF was as high as 46.2%.Compared with baseline, up to 70% of subjects achieved a relative reduction in liver fat of ≥30%.The summary of MRI-PDFF results is shown in the figure below:Image Source: Corporate Official WebsiteTreatment with ALG-055009 significantly reduced atherosclerotic lipids, including LDL-C, lipoprotein (a) (LpA), and apolipoprotein B (ApoB). Additionally, a dose-dependent increase in sex hormone-binding globulin (SHBG) was observed, which is a marker of THR-β targeting in the liver.ALG-055009 is a potential best-in-class THR-β agonist developed by Aligos Therapeutics. In March 2024, the world's first innovative MASH drug, Resmetirom from Roche, which also targets THR-β, was approved by the FDA. However, this drug has not yet been approved in China.
2. Daiichi Sankyo/MSD: HER3 ADC Meets Clinical Endpoint, Planned for Market SubmissionOn September 17, Daiichi Sankyo and Merck jointly announced that the HER3 ADC drug patritumab deruxtecan (HER3-DXd) demonstrated a statistically significant improvement in PFS (the primary endpoint of the trial) in the HERTHENA-Lung02 Phase III clinical study. This study evaluated the efficacy of the drug in EGFR-mutated NSCLC patients previously treated with frontline EGFR-TKI therapy. OS data are not yet mature and will continue to be assessed.Daiichi Sankyo stated that the HERTHENA-Lung02 study data will be presented at upcoming medical conferences and shared with global regulatory authorities to discuss the next steps. This may indicate that the FIC new drug is approaching its market application.HER3-DXd is the third ADC "masterpiece" in Daiichi Sankyo's pipeline, following Enhertu and Dato-DXd.In terms of progress, it is far ahead among ADCs with the same target;In terms of efficacy, previous data has also demonstrated its effectiveness in EGFR-mutant NSCLC, potentially offering a new treatment option for a broad patient population with disease progression after EGFR-TKI therapy or with various resistance mutations.HERTHENA-Lung02 is an open-label, global, multicenter Phase III clinical trial comparing the efficacy and safety of HER3-DXd (5.6mg/kg Q3W) versus chemotherapy (pemetrexed + cisplatin) for four cycles in patients with EGFR-mutated locally advanced or metastatic NSCLC who have failed third-generation EGFR-TKI treatment. The primary endpoint is PFS assessed by BICR, and secondary endpoints include OS, ORR, DoR, etc.3. Roche: New Phase 3 Clinical Trial of Baloxavir Marboxil Meets Primary Endpoint
On September 19, Roche announced positive results from the CENTERSTONE Phase III clinical study. This clinical trial evaluated the impact of baloxavir marboxil (Xofluza) on the household transmission of influenza in respiratory viral diseases, enrolling over 4,000 participants. The results showed that a single oral dose of Xofluza significantly reduced the likelihood of household members contracting the influenza virus from an infected individual.
Baloxavir MarboxilIs a new anti-influenza drug, which was initiallyDeveloped by Japanese pharmaceutical company Shionogi. In 2016, Roche reached a cooperation agreement with Shionogi to jointly take charge of the drug's research and development outside Japan and Taiwan, with Roche holding commercial promotion rights in regions other than Japan and Taiwan.In April 2021, it was approved by the NMPA for marketing in China under the trade name Xofluza.Xofluza is a single-dose oral medication with a completely new mechanism of action against influenza., This drug belongs to the Cap-dependent endonuclease inhibitor, which aims to inhibit the CAP cap structure-dependent endonuclease in the influenza virus. This enzyme is essential for the replication of the influenza virus. Notably, this drug is the first anti-influenza medication approved in the United States in nearly 20 years and is Roche's successor toAnother Heavyweight Anti-Influenza Drug Following Tamiflu (Oseltamivir)。
The currently widely used oral anti-influenza medication is the neuraminidase inhibitor class of antiviral drugs (oseltamivir), which requires twice-daily dosing for five consecutive days. In contrast, baloxavir marboxil (Xofluza), with a novel mechanism of action, directly inhibits viral replication. Patients need only take it once during the entire course of treatment to stop viral shedding within 24 hours.
Progress of Innovative Drugs in ChinaThis week, a total of 55 innovative drugs (including improved new drugs) in China have advanced to new stages of development, with 4 submitting for marketing approval, 3 initiating Phase III clinical trials, 23 new drugs receiving clinical approval, and 11 applying for clinical trials.
Four Innovative Drugs (Including Modified New Ones) Approved for Clinical Trials in China for the First Time This Week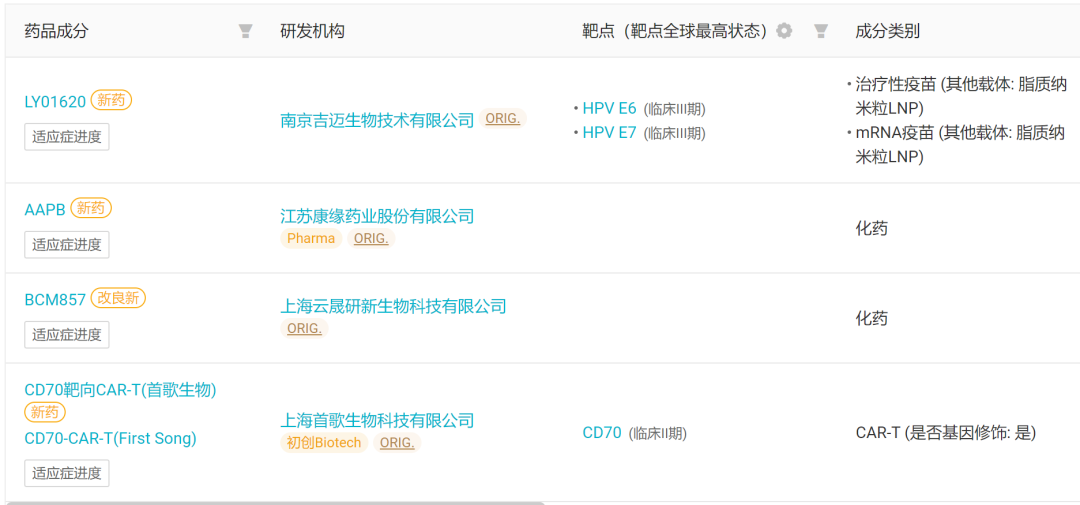
Source: Insight Database Web Version(The following text is from the same source unless otherwise specified.)1. Hengrui Medicine: The World's First PD-L1/TGF-βRII Bifunctional Fusion Protein Submitted for Marketing Approval, First-Line Treatment for Gastric CancerOn September 19, Hengrui Medicine released a press release stating that its submitted Class 1 new drugAnti-PD-L1/TGF-βRII Bifunctional Fusion Protein「Relaforp-α Injection (SHR-1701)" has been accepted by NMPA for marketing application, indicated for use in combination with fluorouracil and platinum-based drugs for locally advanced unresectable, recurrent, or metastaticFirst-line Treatment for Gastric and Gastroesophageal Junction Adenocarcinoma. This is also the follow-up toHER2 ADCLater, Hengrui's another Class 1 new drug applied for marketing this month.
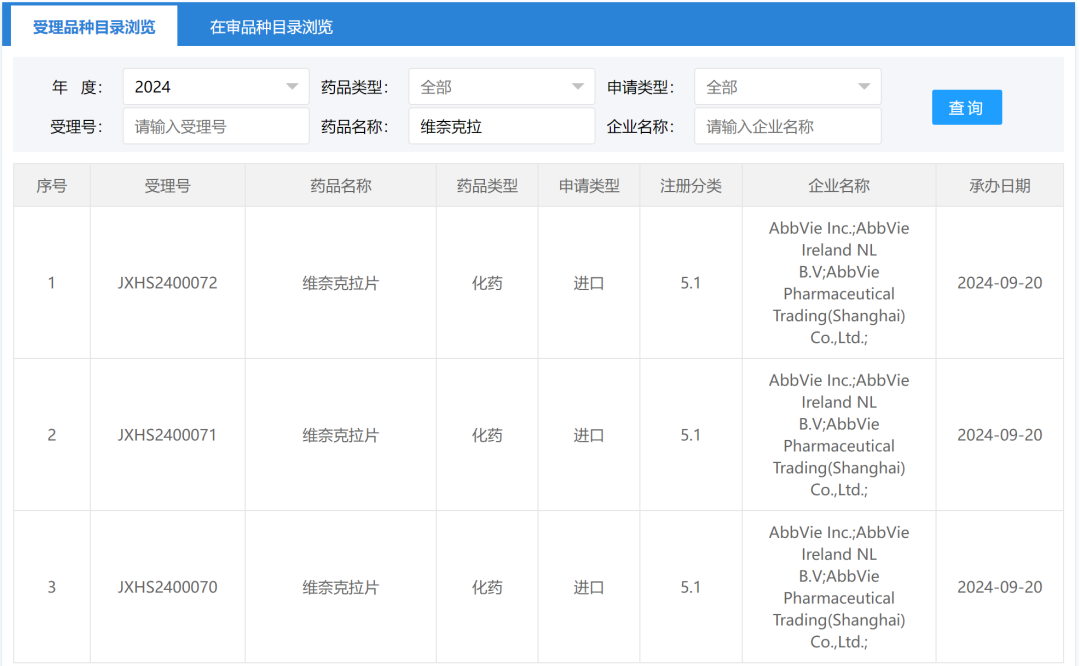
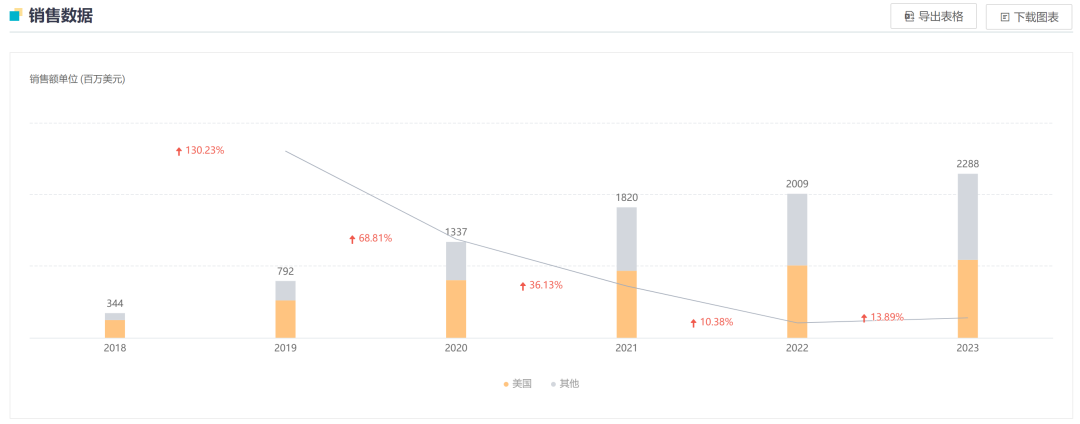
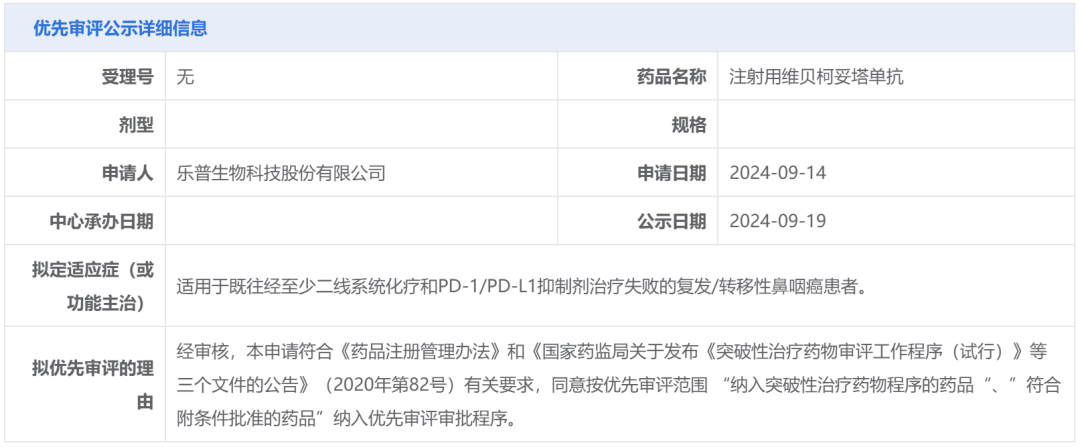
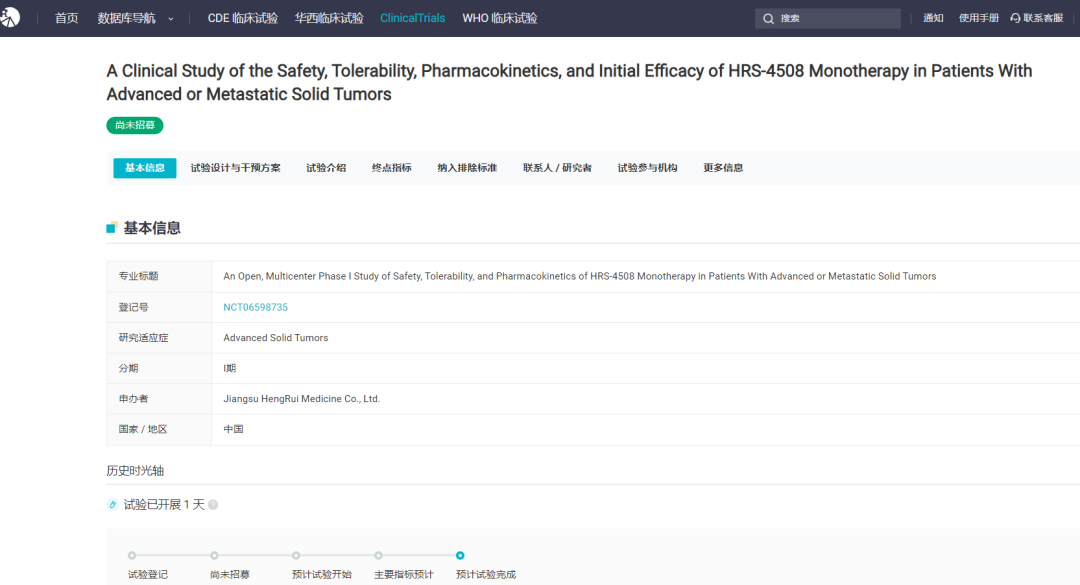
Screenshot source: Hengrui Official WeChatRelafusp-α Injection is a bifunctional fusion protein independently developed by Hengrui Medicine that targets PD-L1/TGF-βRII. It can promote the activation of effector T cells and effectively enhance immune modulation within the tumor microenvironment, ultimately boosting the immune system's ability to kill tumor cells. Currently, no similar products have been approved for marketing in China or globally.Currently, researchers are conducting clinical studies on the use of Relatlimab-α injection in the treatment of various indications, including gastric or gastroesophageal junction adenocarcinoma, rectal cancer, and non-small cell lung cancer, to evaluate its anti-tumor effects across different types of solid tumors.In June 2024, the Relafusp-α injection reached the pre-specified primary endpoint in the Phase III clinical trial SHR-1701-III-307 for gastric cancer. This is a randomized, double-blind, multi-center Phase III study aimed at evaluating the efficacy and safety of Relafusp-α injection combined with chemotherapy compared to placebo combined with chemotherapy in patients with advanced or metastatic gastric or gastroesophageal junction adenocarcinoma. Professor Lin Shen from Peking University Cancer Hospital served as the principal investigator, with participation from over 70 centers across China. The primary endpoint was overall survival (OS). Secondary endpoints included progression-free survival (PFS), objective response rate (ORR), duration of response (DoR), disease control rate (DCR), and safety.This Phase III study enrolled a total of 737 subjects. The results showed that the Relatlimab-α injection combined with chemotherapy group significantly outperformed the placebo combined with chemotherapy group on the primary endpoint, significantly prolonging the overall survival of patients with advanced gastric or gastroesophageal junction adenocarcinoma. No new safety risk signals were identified, and the safety profile was acceptable. The study results were presented at the 2024 European Society for Medical Oncology (ESMO) Congress.2. Ocumension: New Drug for Post-Cataract Surgery Inflammation Submitted for Market ApprovalOn September 20, the CDE website showed that Ocumension's Class 5.1 imported new drug "Dexamethasone Implant"」Listing Application Accepted(Application No.: JXHS2400069), used for treating post-cataract surgery inflammation.Screenshot source: CDE official websiteDexamethasone Implant (R&D Code:OT-502) is a drug that Ocumension Therapeutics introduced from EyePoint Pharmaceuticals in January 2020. According to the cooperation agreement, EyePoint will receive a prepayment of 2 million US dollars and tiered sales royalties, with additional development, regulatory, and commercial sales milestones totaling up to 12 million US dollars; andOcumension Therapeutics has obtainedThe exclusive rights to develop and commercialize the product in mainland China, Hong Kong, Macao, and Taiwan.OT-502 is a novel, biodegradable, single-injection sustained-release anti-inflammatory drug that immediately releases the active ingredient dexamethasone upon injection to exert an anti-inflammatory effect, with continuous release over 21-22 days. Being biodegradable, it does not require removal. OT-502 utilizes Verisome drug delivery technology to deliver dexamethasone to the anterior chamber, directly inhibiting the synthesis and release of inflammatory mediators within the anterior chamber. It provides continuous and stable suppression of anterior chamber inflammation post-surgery, avoiding the frequent use of steroid eye drops. This effectively addresses the serious issue of poor postoperative medication adherence in some cataract patients, offering a better option for managing post-cataract surgical inflammation.On April 15, 2024, Ocumension announced that OT-502, a new drug for treating postoperative inflammation, has completed Phase III clinical trials in China. The randomized, double-blind, placebo-controlled, parallel-group, multicenter Phase III clinical trial evaluated the efficacy and safety of a 9% dexamethasone implant for treating inflammation after cataract surgery. The results showed that OT-502 met the pre-specified primary efficacy endpoint, with a significantly higher proportion of participants achieving a grade 0 anterior chamber cell score on day 8 compared to the placebo group, demonstrating the product's safety and effectiveness in controlling post-cataract surgery inflammation.Ocumension currently has 33 drug assets for anterior and posterior segments of the eye, establishing a complete ophthalmic drug product line, with 20 products at the commercialization stage, 3 products in Phase III clinical trials, and 2 products entering the commercial registration stage.3. AbbVie: Bcl-2 Inhibitor "Venetoclax" New Indication Submitted for Market ApprovalOn September 20, the CDE website showed that AbbVie's Bcl-2 inhibitor "Venetoclax" new indication was submitted for marketing.(Application No.: JXHS2400070/1/2)。This is the third marketing application for the drug in China, and the indication has not been disclosed yet.Screenshot source: CDE official websiteVenetoclax (ABT-199), co-developed by AbbVie and Roche, is an oral small-molecule targeted drug that inhibits Bcl-2. By binding to and inhibiting Bcl-2, it releases pro-apoptotic proteins, thereby killing cancer cells.Bcl-2 is one of the key proteins regulating apoptosis, overexpressed in various human tumor tissues and cells, and closely related to the occurrence, development, and drug resistance of tumors.In April 2016, Venetoclax was approved by the U.S. FDA for the treatment of chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), becomingThe world's first approved Bcl-2 inhibitor;December 2020, Venetoclax for the first timeIn ChinaApproved for marketing, to be used in combination with azacitidine for the treatment of patients who are not suitable for intensive induction chemotherapy due to comorbidities or age.Newly diagnosed adult AML patients aged 75 years and above. Currently, there are two marketing applications for this drug under review in China, which are worth anticipating.According to Insight DataAccording to the database, the global sales of Venetoclax reached 2.288 billion US dollars in 2023, maintaining steady growth.1. Lepu Biotech: EGFR ADC Proposed for Priority ReviewOn September 19, the CDE website showed that Lepu Biopharma's EGFR ADC new drug, Vericetuzumab (MRG003), is proposed to be included in the priority review.For patients who have previously failed at least two lines of systemic chemotherapy and PD-1/PD-L1 inhibitor treatment.Patients with Recurrent/Metastatic Nasopharyngeal Carcinoma。According to the Insight database, currently, there is no EGFR-targeted ADC approved for marketing worldwide. Lepu is expected to take the lead and become the first.The World's First。Image Source: CDE Official WebsiteMRG003 is an ADC composed of an EGFR-targeted monoclonal antibody conjugated with a potent microtubule inhibitor payload, the MMAE molecule, via a vc linker. The indication proposed for priority review has previously been granted the status of a breakthrough therapy by the CDE in China, while overseas, the U.S. FDA has also awarded MRG003 orphan drug designation, fast track designation, and breakthrough therapy designation for the treatment of recurrent or metastatic nasopharyngeal carcinoma.At the 2024 ASCO Annual Meeting, LePu Bio announced the Phase I/II study on the safety and efficacy of MRG003 in combination with the PD-1 inhibitor Putelimab for treating patients with EGFR-positive solid tumors. As of January 30, 2024, 33 patients were enrolled, with the Phase I study including 9 nasopharyngeal cancer patients, 1 head and neck squamous cell carcinoma patient, and 3 patients with other solid tumors, while the Phase II portion included 14 nasopharyngeal cancer patients and 6 head and neck squamous cell carcinoma patients.The test data showed,InAmong 27 evaluable patients, 17 patients achieved PR, 7 patients achieved SD, with an ORR of 63.0% and a DCR of 88.9%. In the Phase II clinical trial, among 9 evaluable EGFR-positive nasopharyngeal carcinoma patients who received first-line treatment with PD-1 plus platinum-based chemotherapy, 2 patients achieved complete response (CR), 5 patients achieved PR, and 2 patients achieved SD, with an ORR of 77.8% and a DCR of 100%.Insight database shows that the two fastest-progressing products in the EGFR ADC field are both from China. Apart from Lepu's MRG003, CPO301 from CSPC Pharmaceutical Group is the second most advanced product, having already entered Phase III clinical trials in China. Additionally, multiple EGFR ADCs have entered clinical trials in China, including BB-1705 from BrightGene Bio-Medical, HLX42 from Henlius/Yilian Biotechnology, and DXC004A from Dox Bio.2. CANbridge: Gaucher Disease Type 1 New Drug Granted Priority ReviewOn September 20, the CDE website showed that北海康成's enzyme replacement therapy "Velaglucerase Alfa for Injection" (CAN103) is proposed to be included in the priority review, applicable toLong-term Enzyme Replacement Therapy (ERT) for Patients Diagnosed with Gaucher Disease Type I (GD1) and Type III (GD3)。Image Source: CDE Official WebsiteCAN103 is a treatment for Type I and Type III Gaucher disease patients developed by Canbridge.Recombinant Human Cerebroside Enzyme Replacement TherapyMost patients with Gaucher disease have type I and type III, which are chronic non-neuropathic and chronic neuropathic types, respectively. CAN103 specifically supplements the deficient glucocerebrosidase in the lysosomes of patients with Gaucher disease through intravenous infusion.On August 19,北海康成 announced positive topline results from the pivotal clinical trial of CAN103 in treatment-naïve subjects aged 12 years and older with Type I and Type III Gaucher disease.The pivotal clinical trial of CAN103 is a randomized, double-blind, dose-comparison study designed to evaluate the efficacy, safety, and pharmacokinetics of intravenous infusion of CAN103 every two weeks in treatment-naïve Gaucher disease patients, with an open-label extension phase. The results indicate that the study successfully met its primary efficacy endpoint in both the 60U/kg dose group (P<0.0001) and the lower 30U/kg dose group (P<0.001), which is the mean percentage reduction in spleen volume from baseline after 9 months of treatment.All key secondary efficacy endpoints, namely, the reduction in liver volume compared to baseline, increase in hemoglobin levels, and rise in platelet counts in the 60U/kg dose group, also achieved statistically significant improvement.It is reported that CAN103 is the first collaboration project between Canbridge and Wuxi Biologics in the field of rare diseases and is China's first enzyme replacement therapy (ERT) developed for Gaucher's disease. CAN103 aims to provide long-term treatment for adult and pediatric patients with Type I and Type III Gaucher's disease. According to Frost & Sullivan, there were approximately 3,000 patients with Gaucher's disease in China in 2020. Launch Phase III Clinical Trial1. Hengrui Medicine: Parkinson's Disease New Drug Initiates Phase III Clinical TrialOn September 19, Hengrui registered a Phase III clinical trial on the Clinicaltrials website to evaluate the efficacy and safety of HRG2010 in treating Parkinson's disease.(NCT06598735)。Image Source: Insight DatabaseHRG2010 Capsule is a novel compound sustained-release capsule of Carbidopa and Levodopa independently developed by Hengrui. This Phase 3 clinical trial...Using carbidopa-levodopa sustained-release tablets as a control, the primary endpoint was the change in "off" time at week 21 compared to baseline.Parkinson's disease is a common neurodegenerative disorder in middle-aged and elderly individuals, characterized pathologically by damage to the dopaminergic neurons of the nigrostriatal pathway in the central nervous system, with clinical features including resting tremor, bradykinesia, muscular rigidity, and ataxia.Currently, similar products that have been marketed both in China and internationally mainly include Levodopa and Benserazide Hydrochloride Tablets (trade name: Madopar), Entacapone Bilastine Tablets (trade name: Stalevo), and Carbidopa and Levodopa Controlled-Release Tablets (trade name: Sinemet).According to the Insight database, generic versions of Carbidopa and Levodopa Extended-Release Tablets have already been approved for marketing in China. In addition, several others are currently applying for market approval. Currently, only Hengrui offers capsules.2、AbbVie: CD20×CD3 Bispecific Antibody Phase III Clinical Trial Initiated in China, First-Line Treatment for Follicular Lymphoma
On September 19, the Drug Clinical Trial Registration and Information Disclosure Platform showed that AbbVie registered a Phase III clinical trial in China to evaluate the safety and efficacy of Epcoritamab + Rituximab and Lenalidomide compared with chemoimmunotherapy in previously untreated follicular lymphoma subjects.Epcoritamab is the third CD20×CD3 bispecific antibody approved globally and one of the CD20×CD3 bispecific antibody products with faster research progress in China. The drug was listed by the industry media Evaluate as one of the top ten potential blockbuster therapies in 2023.
Screenshot from:Drug Clinical Trial Registry and Information Disclosure PlatformEpcoritamabInitially developed by Genmab, AbbVie entered into a collaboration with Genmab in 2020 worth up to $3.9 billion to co-develop and commercialize three of Genmab's next-generation bispecific antibody products, including Epcoritamab.Epcoritamab is produced using Genmab's proprietary DuoBody technology. The company’s DuoBody-CD3 technology is designed to selectively direct cytotoxic T cells toward tumors to induce an immune response against malignant tumor cells. As a bispecific antibody, Epcoritamab can simultaneously bind to CD3 on T cells and CD20 on B cells, inducing T cell-mediated killing of lymphoma B cells.
Epcoritamab Global First Approval on May 19, 2023: FDA Approval for High-Grade B-Cell Lymphoma (Third-Line) and Diffuse Large B-Cell Lymphoma (DLBCL)Since 2024, Epcoritamab has gained new indications. In June and August this year, the drug received FDA and EMA approval respectively for the treatment of follicular lymphoma (third-line, last-line). According to AbbVie's financial report, global sales of Epcoritamab reached $63 million in the first half of 2024. With the expansion of new indications, Epcoritamab’s sales this year are expected to exceed $100 million without any problem.Epcoritamab Research Historical Timeline
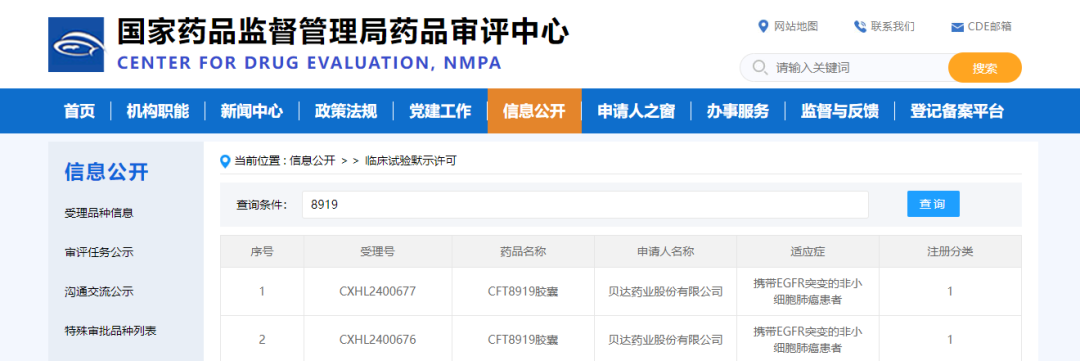
Screenshot source: Insight DatabaseIn China, Epcoritamab has not yet been approved. Previously, AbbVie and Genmab had initiated three Phase III clinical trials of Epcoritamab in China; the newly registered trial is a multicenter, randomized, open-label study (EPCORE™FL-2,CTR20243120), aiming toPreviously Untreated Follicular LymphomaEvaluation of the Safety and Efficacy of Epcoritamab + Rituximab and Lenalidomide versus Chemoimmunotherapy in Subjects. The study plans to enroll 162 participants in China and 1080 participants internationally. Approved for Clinical Use1. Betta Pharmaceuticals: PROTAC New Drug Approved for Clinical Trials, Deepening Lung Cancer Field LayoutOn September 19, the CFT8919 capsule co-developed by Betta Pharmaceuticals and C4 Therapeutics, Inc. (C4T) received the "Clinical Trial Approval Notice" (Notice No.: 2024LP02116, 2024LP02117) issued by the National Medical Products Administration, intended for use in non-small cell lung cancer (NSCLC) patients with EGFR mutations.Source of the image: CDE official websiteCFT8919 is an allosteric BiDAC™ (bifunctional protein) degrader with oral bioavailability. In preclinical studies, CFT8919 demonstrated activity in both in vitro and in vivo models of EGFR L858R-driven NSCLC, targeting a broad range of on-target resistance mutations and showing intracranial activity, with the potential to prevent or treat brain metastases in patients. By binding to the allosteric site of the L858R mutation, CFT8919 exhibits strong selectivity while remaining effective against secondary EGFR resistance mutations such as T790M or C797S. In 2023, CFT8919 tablets received Investigational New Drug (IND) approval from the U.S. FDA.Subgroup analyses of several large randomized controlled trials have shown that first-, second-, and third-generation EGFR-TKIs demonstrate varying efficacies in treating patients with 19del and 21L858R mutations. Overall, patients with the 19del mutation benefit significantly more than those with the 21L858R mutation. For NSCLC patients carrying the 21L858R mutation, there remains a substantial unmet clinical need, and better treatment options are eagerly anticipated.On May 29, 2023, Betta announced a collaboration with C4 Therapeutics, Inc. to acquire the exclusive rights for the development, manufacturing, and commercialization of this project in the Greater China region, as well as a specified percentage of sales royalties outside the aforementioned region. The deal involves an upfront payment of $10 million, potential milestone payments of up to $357 million, and sales royalties within the licensed territory. Additionally, Betta plans to subscribe to 5,567,928 newly issued ordinary shares of C4 Therapeutics for $25 million through its wholly-owned subsidiary, Betta Investment (Hong Kong) Co., Ltd.Source:Official News/Information Released by Pharmaceutical Companies, Insight DatabaseCover Source:ZCool Hello Plus
Disclaimer:This article is for information sharing only, and does not represent the position or viewpoint of Insight. It does not recommend or introduce any treatment plans. If you have any needs, please consult and contact正规医疗机构.SubmissionWeChat: insightxb; Email: insight@dxy.cn
Click the card to enterInsight Mini ProgramReview Progress in China, Global New Drug Development…Diversified functions, traceable data……Insight Database Web Version Awaits Your ExperienceClick to read the original text,ImmediatelyUnlock!