Amgen Discloses Discovery and Pharmacological Profile of AMG 193, a Second-Generation MTA-Cooperative PRMT5 Inhibitor for MTAP-Deficient Cancers

Amgen
Developer of Treatment Drugs for Serious Diseases
1. Background Introduction
Synthetic lethality can be used to target and treat tumors with mutations, meaning that when cancer cells can individually tolerate disturbances in either gene without phenotypic effects, but lose viability when both genes are perturbed simultaneously, a synthetic lethal interaction occurs between the two genes. The clinical validation of synthetic lethality has been achieved through the approval of various PARP inhibitors for the treatment of BRCA1- and BRCA2-mutated cancers. In many tumors, tumor suppressorsCDKN2AAnd the nearby S-methyl-5-thioadenosine phosphorylase (MTAP) Gene co-deletion, this type of cancer cell is sensitive toProteinArginine Methyltransferase 5(PRMT5) significant dependence. Approximately 10-15% of tumors exhibit genomic loss at this locus, particularly in tumors with unmet clinical needs such as pancreatic cancer, lung cancer, and glioblastoma.MTAPPlays a central role in the adenine and methionine salvage pathways, and the loss of MTAP leads to the metabolite 5-methylthioadenosine (MTA) Accumulation.MTA is an inhibitory PRMT5 cofactor that competes with the methyl donor S-adenosylmethionine (SAM) for binding to PRMT5.Therefore, in MTAP-deficient cancer cells, the activity of PRMT5 is partially inhibited, making these cells particularly vulnerable to further PRMT5 inhibition.
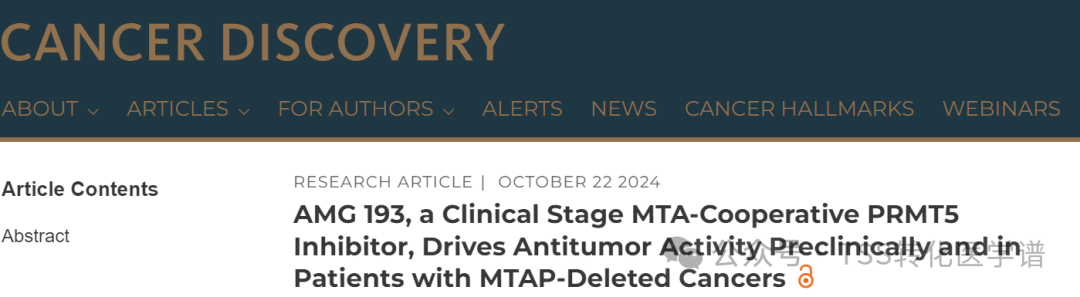
Methyltransferase PRMT5 in Cells andMEP50Forming complexes that catalyze the symmetric dimethylation of arginine residues on many substrates, including transcription factors, histones, and members of the RNA splicing complex. Structurally,Four PRMT5 and four MEP50 form a hetero-octameric complex.In this complex, the PRMT5 molecules form two dimers in a conserved head-to-tail arrangement, while the PRMT5 tetramer forms the core of the complex. MEP50 binds to PRMT5 through the N-terminal TIM barrel. Research shows,Compared with PRMT5 alone, the complex of PRMT5 and MEP50 has higher methyltransferase activity.This may be due to the metastable state of MEP50 in cofactor and protein binding. The PRMT5 enzyme catalyzes the methylation of non-histone substrates and thus plays a role in various important cellular functions.
In addition, PRMT5 is upregulated in many cancers and is known to play a role in driving oncogenic growth, making it an attractive therapeutic target. Multiple SAM-competitive or -cooperative PRMT5 inhibitors have been described, some of which have entered clinical stages (PF-06939999,GSK3326595,JNJ-64619178). However, the activity of these molecules is not limited to MTAP-deficient cells, and in MTAP wild-type cells, a significant mechanism was observed due to PRMT5 inhibition.Hematological ToxicityIt is speculated that in the presence of MTA, a small molecule that specifically binds to and inhibits PRMT5 may selectively target MTAP-deficient cancer cells while preserving PRMT5 activity in MTAP wild-type (WT) cells.AMG 193It is a second-generation PRMT5 inhibitor and a PRMT5-MTA complex inhibitor.
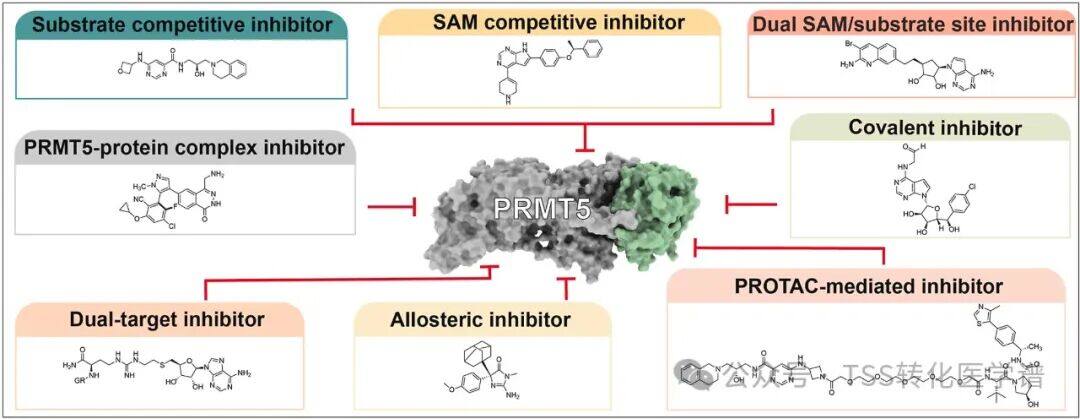
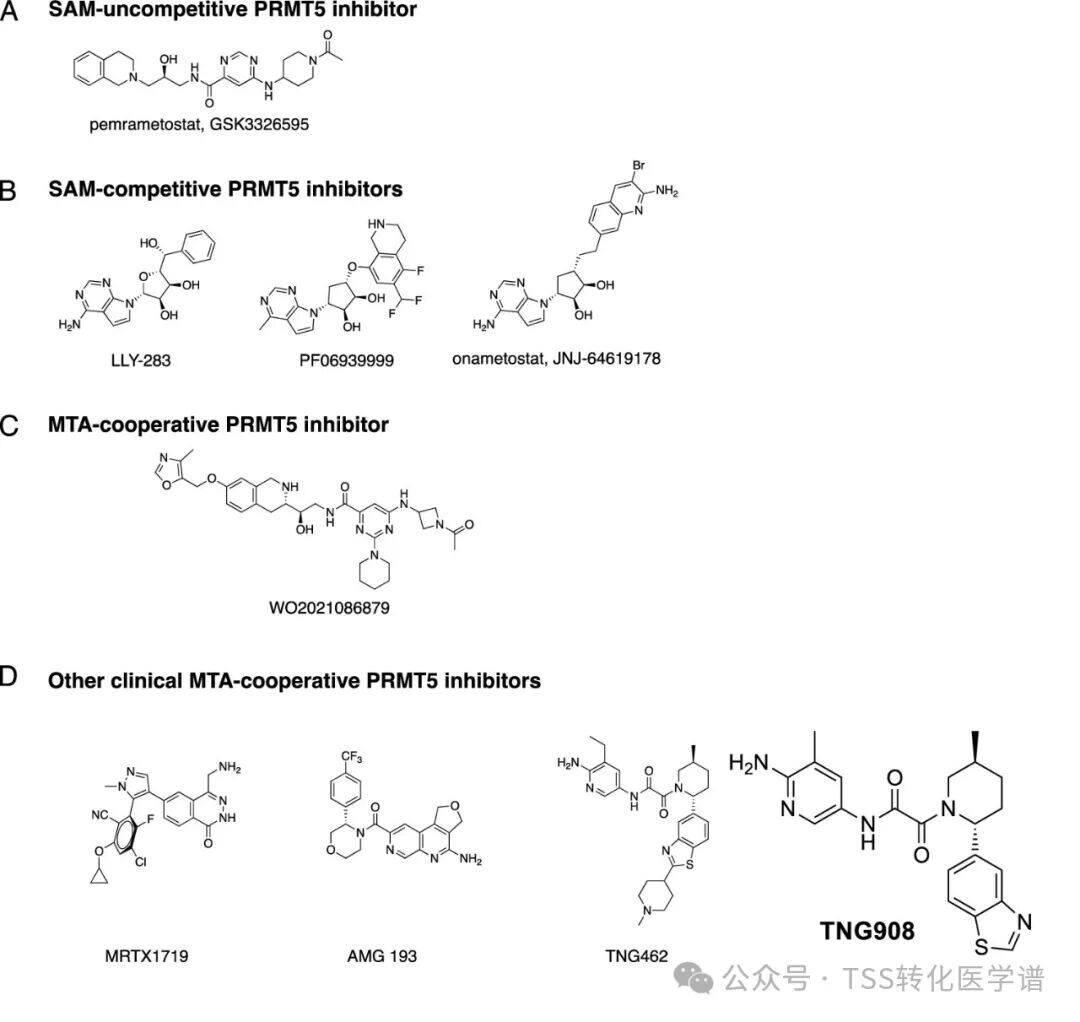
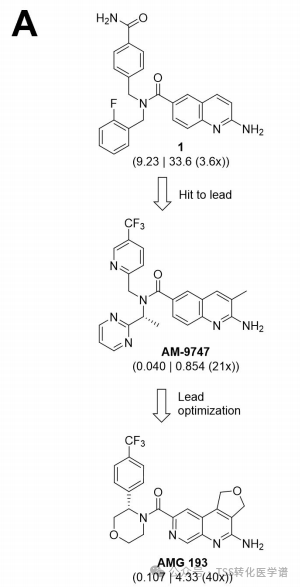
2. Molecular Discovery of AMG 193, a Potent MTA-Cooperative PRMT5 Inhibitor
To identify small molecules that preferentially bind to the PRMT5-MTA complex in the presence of MTA, the authors screened a DNA-encoded library targeting PRMT5 (DELs): PRMT5:MEP50 (6µM) in the presence of MTA (60µM) or Sinefungin (60µM, SAM analog), containing98.4 millionA compound. This screening discoveredAminoquinoline Compound 1(Figure 1,aminoquinoline compound 1) as the initial hit compound targeting PRMT5-MTA.Compound 1The half-maximal inhibitory concentration (IC50) in HCT116 MTAP-deficient cells50 9.23µM and 3.6xSelective (compared to HCT116 MTAP WT cells). Further optimization yielded the compound.AM- 9747,Its intracellular potency (MTAP-deficient cells,IC50=0.040µM) and selectivity (21x)。AM-9747It demonstrated certain pharmacokinetic activity (mouse intravenous clearance 2.3 L/h/kg) and oral bioavailability (%F=23), making it suitable for in vivo proof-of-concept studies. Subsequent lead compound optimization using structure-based and drug-likeness property-based drug design led to the discovery of tricyclic amide compounds.AMG 193, it is effective (with a half-maximal inhibitory concentration in MTAP-deficient cells of0.107µM), MTA-assisted selectivity (40x) and orally bioavailable in preclinical species (100% in dogs).
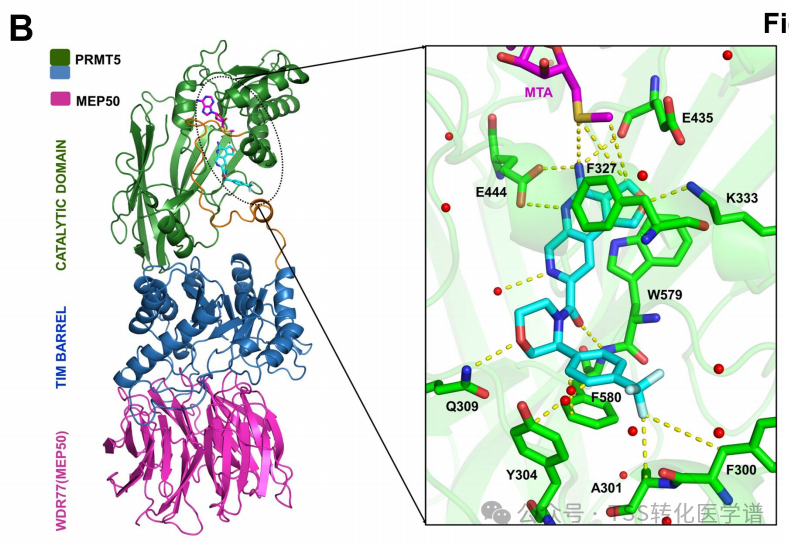
The X-ray co-crystal structure of AMG 193 in the MTA-bound PRMT5:MEP50 active site was determined (Figure 1B). The structure shows that AMG 193 occupies the substrate-binding pocket of PRMT5, interacting withGlu444The side chain forms strong polar interactions, with the sulfur atom of MTA interacting with the amino group of AMG 193. Additionally, the dihydrofuran ring exhibits π-π stacking interactions with the side chains of Trp579 and Phe327, while the O-atom forms a hydrogen bond with the amino side chain of Lys333. This side chain typically interacts with the carboxyl end of SAM, and the binding of AMG 193 causes Lys333 to move away from the binding site of SAM. This series of contacts contributes to the observed selectivity and MTA synergistic inhibition of AMG 193 (Figure 1B). This structure illustrates the trimeric complex formed between AMG 193, MTA, and PRMT5; thus, AMG 193 is considered a PRMT5 inhibitor that cooperates with MTA.
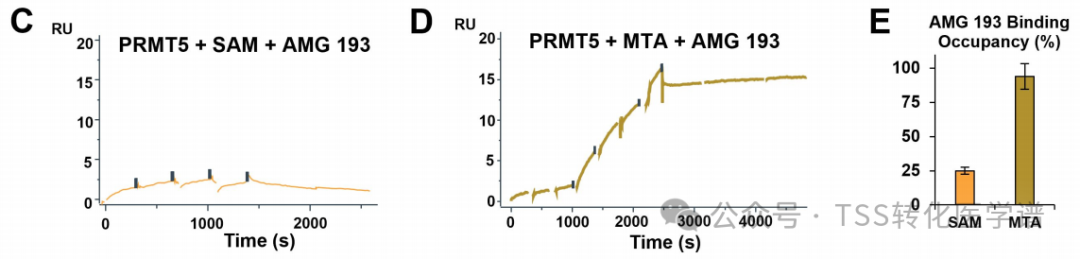
In the presence of MTA and SAM, the binding affinity (KD) and kinetic parameters (association rate (ka) and dissociation rate (kd))), in vitro half-life (t1/2) and the synergy of MTA. Analysis of direct SPR binding shows,In the presence of MTA, AMG 193 forms a highly stable complex with PRMT5. The dissociation rate of the ternary complex is very slow, with a kd of 1.0.-4 1/s(t1/2>120 min)。In contrast,AMG 193 binds to the PRMT5-SAM complex in a less effective manner,Dissociation rate is faster, and the half-life is shorter (t1/2<30 min), with weaker binding affinity (KD = 0.23 nM) (Figure 1C). Compared with MTA, this reduction in affinity leads toThe binding occupancy percentage of AMG 193 is lower in the presence of SAM.(about 25% vs. 94%). AMG 193 exhibits a slow dissociation rate from the PRMT5-MTA complex, which hinders the direct measurement of accurate binding and dissociation rate constants. Therefore, Amgen designed a tracer molecule (Compound 2, AMG193 competitive molecules,chaser molecule, the SPR method for determining slow dissociation rate constants, see: 10.1016/j.ab.2024.115679), this is a PRMT5 binder independent of MTA, with an affinity of 6 nM (± MTA) and a short half-life (t1/2<10min)(Figure 1D), AMG193 binds with extremely high affinity to the PRMT5-MTA complex (3.9 mM), generating a stable complex dissociation rate (kd = 7.78-61/s and t1/2Approximately 25 hours) and high binding surface occupancy of approximately 94% (Figures 1D and 1E). The cooperativity of AMG 193 was calculated by comparing the SPR affinity of AMG 193 with PRMT5-MTA and PRMT5-SAM complexes. Overall, inIn the presence of MTA, the binding ability of AMG 193 to PRMT5 is approximately 60 times higher, with a binding surface occupancy increased by about 70%, forming a highly stable complex with an extremely slow dissociation rate. In summary,SPR confirmed that AMG 193 is an MTA co-binder and PRMT5 inhibitor.
3. Tumor cells with MTAP deletion exhibit increased sensitivity to MTA-cooperative PRMT5 inhibitors.
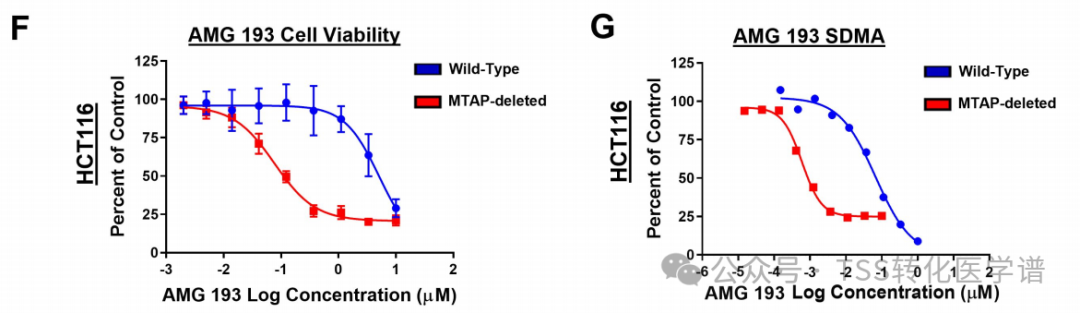
To investigate the effects of MTA-cooperative PRMT5 inhibitors on cancer cell viability and signal transduction, MTAP-WT and MTAP-deficient HCT116 cells were used. A dose-dependent decrease in cell viability was observed in both WT and MTAP-deficient cells. However, MTAP-deficient cells were more sensitive to MTA-cooperative PRMT5 inhibitors, with lower half-maximal inhibitory concentration values.46 Times(Figure 1F). The overall symmetric dimethylarginine (SDMA) levels also decreased in a dose-dependent manner, with the half-maximal inhibitory concentration (IC50) value of MTAP-deficient cells reduced by 90-fold (Figure 1G). MTA-coordinated PRMT5 inhibitorAMG 193 and AM-9747Can preferentially suppress SDMA levels and cell viability in MTAP-deficient cells. AMG 193 has entered clinical studies due to improved in vivo efficacy and PK properties.
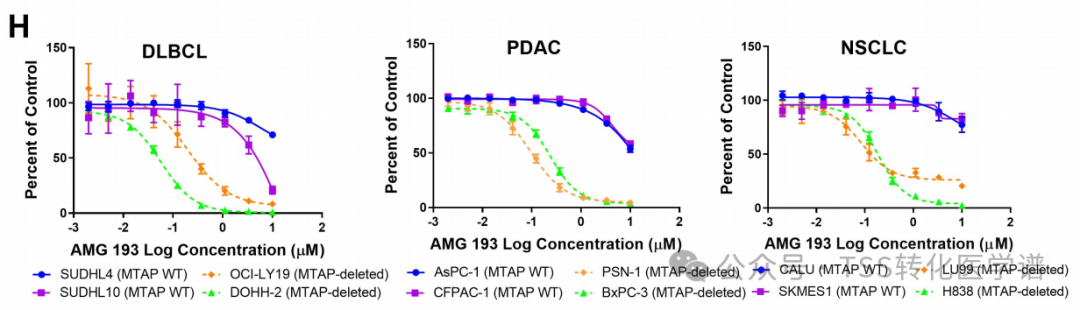
MTA-cooperative PRMT5 inhibitor AM-9747 or non-cooperative PRMT5 inhibitor was tested in a panel of cancer cell lines with endogenous MTAP WT and MTAP deletion.LLY-283In tumor types with MTAP deficiency, there is a clear separation between WT and MTAP-deficient lines, including diffuse large B-cell lymphoma (DLBCL), pancreatic ductal adenocarcinoma (PDAC), and non-small cell lung cancer (NSCLC).Figure 1H). For most WT cell lines, the accurate half-maximal inhibitory concentration (IC50) value could not be calculated as the curve did not reach it. Notably, MTAP-deficient DLBCL cell lines are highly sensitive to PRMT5 inhibition, with an IC50 value 10 times lower than other indications (Figure 1H)。
4. MTA-Cooperative PRMT5 Inhibition Induces Selective Splicing Defects, Cell Cycle Arrest, and DNA Damage Response in MTAP-Deficient Cells
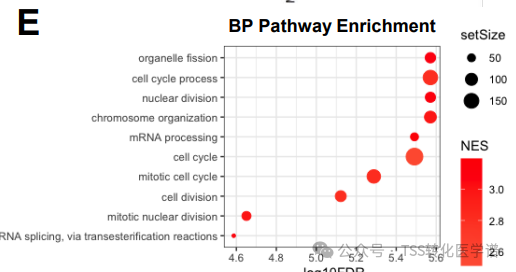
To investigate the mechanism of action (MOA) of MTA-cooperative PRMT5 inhibitors, RNA-seq was used to evaluate global gene expression changes induced by PRMT5 inhibition. Gene set enrichment analysis (GSEA) revealed that MTA-cooperative PRMT5 inhibitors affect cell growth by altering cell cycle gene expression and increasing alternative mRNA splicing.
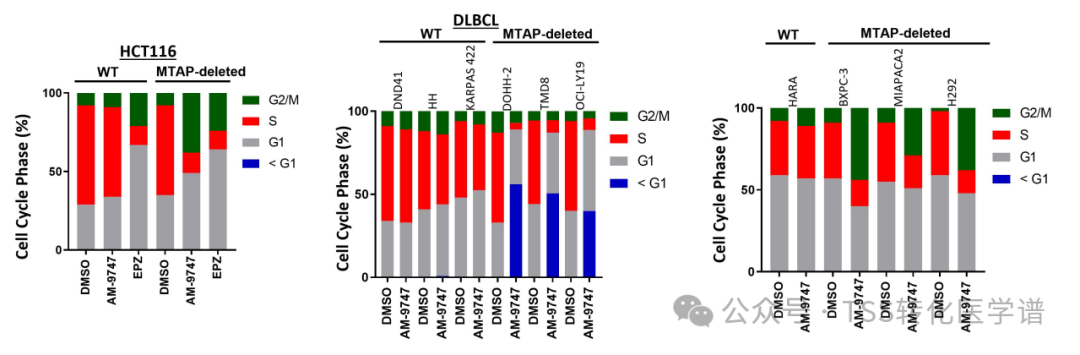
To investigate the effect of PRMT5 inhibition on the cell cycle, paired isogenic HCT116 cell lines were treated with a PRMT5 inhibitor and analyzed for cell cycle distribution by flow cytometry. Compared with WT cells, AM-9747 treatment showed a selective reduction in S phase cells and a selective increase in G2/M phase cells in MTAP-deficient cells.Figure 2A). In contrast, using non-cooperative PRMT5 inhibitorsEPZ015666(EPZ)After treatment, similar phenotypes were observed in both WT and MTAP-deficient cells. In a set of DLBCL WT and MTAP-deficient cells, AM-9747 treatment selectively induced cell death, which could be attributed solely to the MTAP-deficient cells.
5. Whole-cell lineage screening confirmedAMG 193Sensitivity Associated with MTAP Loss
To further evaluate the sensitivity and synergy of MTA in combination with the PRMT5 inhibitor AMG 193, a large unbiased screen was conducted, including over 850 cancer cell lines across various cancer indications.(PRISM Platform).Barcoded cancer cell lines were treated with 8-point dose-response AMG 193 for 5 days, and cell line sensitivity profiles were generated from the area under the curve (AUC) values using relative barcode abundance. The sensitivity profile of AMG 193 showedMTAP LossCell lines are associated with sensitivity to AMG 193 (Fig. 3A-C)。
Using the existing Cancer Dependency Map (DepMap) database, AMG 193 AUCs were correlated with copy number, RNA expression levels, and viability following genome-wide RNAi knockdown (KD) or CRISPR knockout (KO).Figures 3D and 3E). The highest genetic dependency related to the sensitivity of AMG 193 in the RNAi KD dataset is PRMT5, indicating that PRMT5-dependent cell lines exhibit higher sensitivity to MTA-cooperative PRMT5 inhibitors (Fig. 3D, Left). PRMT5 and its binding protein WDR77 are the two most significant associations in the CRISPR KO dataset (Figure 3D, Right). In addition, the highest genomic feature in terms of copy number and RNA expression is MTAP, further demonstrating the selectivity of this compound for MTAP-deficient tumor cells (Figure 3E)
6. AMG 193 Inhibits the Growth of MTAP-Deficient Tumors In Vivo
To evaluate the potency and selectivity of AMG 193 in vivo, HCT116 and other cell line-derived xenograft (CDX) tumor models were studied.AMG 193 Treatment significantly inhibited SDMA concentration in MTAP-deficient tumors; however, >50% SDMA inhibition in MTAP WT tumors was only observed at the maximum dose of 100 mg/kg.Figure 4A)。AMG 193 is also acceptable.Selective InhibitionGrowth of MTAP-Deficient Tumors In Vivo(Fig. 4B-C)。
Further, targeting MTAP-deficient cancer cells via MTA-cooperative PRMT5 inhibition in PDX models can enhance tumor killing while sparing healthy normal cells.。
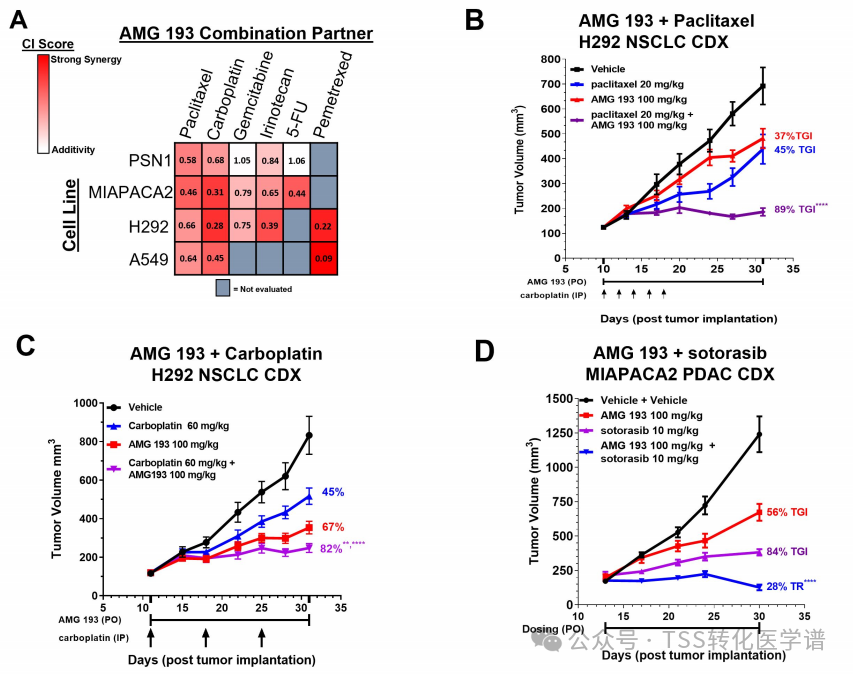
7. AMG 193 Demonstrates Synergistic Effects with Standard Chemotherapy and the KRAS G12C Inhibitor Sotorasib
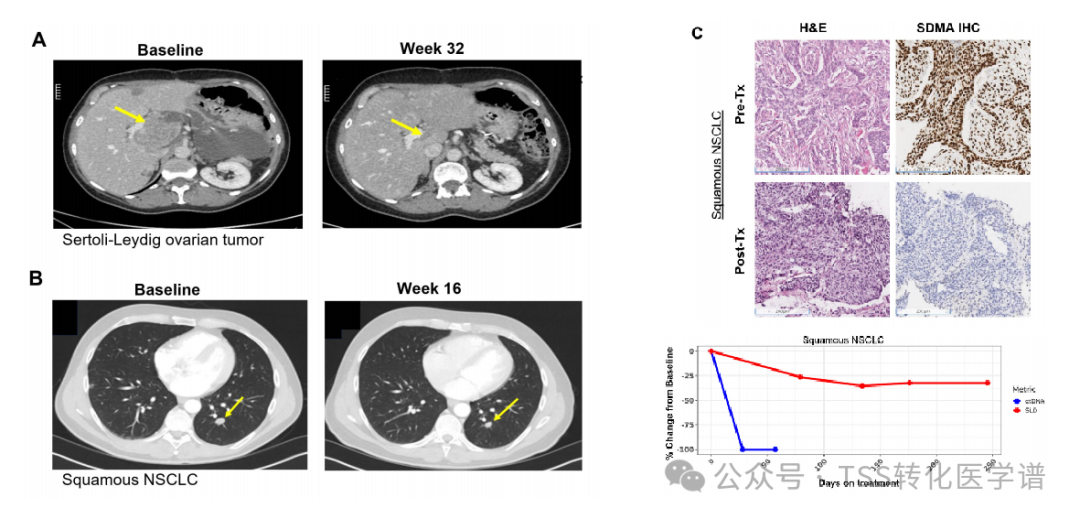
8. AMG 193 Shows Partial Clinical Response in Patients with MTAP-Deleted Solid Tumors
In the ongoing clinical study in human body (NCT05094336), AMG 193 is being evaluated at increasing dose levels in patients with MTAP and/or CDKN2A (MTAP deletion) solid tumors. Clinical activity is assessed every 8 weeks according to the Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Related molecular pharmacodynamic characteristic analyses were performed, including longitudinal monitoring of circulating tumor (ctDNA/cTF) in plasma and SDMA in serum.Cases from responding patients in ongoing clinical trials confirm that there are sufficient relevant biomarkers in these trials to demonstrate the efficacy of AMG 193 in humans.
A 55-year-old woman with ovarian Sertoli-Leydig cell tumor (SLCT), identified with homozygous CDKN2A and MTAP deletions via NGS, was enrolled in clinical trial 20210023. At diagnosis (16 months prior to enrollment), she underwent cytoreductive surgery followed by adjuvant platinum-based chemotherapy, but her disease recurred within 4 months, necessitating peritoneal cytoreduction surgery and further treatment. At the time of enrollment, she had three identifiable target lesions: a liver lesion (79 mm), a perirectal lymph node (50 mm), and a renal lymph node (13 mm). She received AMG 193 800 mg orally (PO) QD in 28-day cycles. Her first disease assessment at week 8 during treatment showed a 13% overall reduction in target lesions, and by week 16, there was a 48% overall reduction, consistent with a partial response as defined by RECIST. Subsequent disease evaluations continued to show gradual decreases in target lesions, reaching a nadir of 80% in cycle 17.Figure 7A). After treatment with AMG 193, serum SDMA decreased within the first week and remained at a lower level.
In addition, one patient received three metastatic treatment regimens, including platinum-based chemotherapy and pembrolizumab. At the time of enrollment, he had multiple pulmonary lesions. He was administered 800 mg AMG 193 PO QD in 28-day cycles. The first disease evaluation post-treatment showed a 26% overall reduction in target lesions, reaching a nadir of 35% at 16 weeks, consistent with a partial response per RECIST criteria (Figure 7B). Tumor tissue biopsy performed in the second cycle showed a 100% reduction in SDMA (Figure 7C). Serum SDMA and ctDNA/cTF decreased within 1 week and 4 weeks after the start of AMG 193 treatment, respectively (Figure 7D). He is still receiving treatment after the 13th cycle, with an overall reduction of 26% compared to baseline, and continues to receive AMG 193 800 mg QD treatment.
Another article was published recentlyAMG 193Phase I clinical trial results, overall ORR only21.4%(n=42, 95%CI, 10.3% to 36.8%)(10.1016/j.annonc.2024.08.2339)。Leading to some skepticism in the market regarding synthetic lethality treatments targeting this site. Competitor company Tanggo (TNG 908/TNG 462) Also fell all the way due to this impact. In addition, Amgen sought toIDE397 (IDEAYA MAT2A Inhibitor) and AMG 193 (Amgen MTA-Cooperative PRMT5 Inhibitor) in Combination Therapy StrategyTumors with MTAP deletion, hoping this combination can address the efficacy issues of this target.
Disclaimer: The publication/reposting of this article is solely for the purpose of information dissemination and does not represent the views of this official account or confirm the authenticity of its content. Any judgment made based on this content will be at your own risk.If there is any infringement, please inform us and we will delete it immediately!
Long press to follow this official account

