J&J Reports Positive Phase 2 Data for Potential Best-in-Class FcRn Antibody Nipocalimab in Moderate-to-Severe Sjögren’s Disease, Demonstrating Over 77% Reduction in IgG

Johnson & Johnson
Healthcare Product Manufacturers, Health Service Providers
Welcome to follow Asymchem Pharma News

On November 14, 2024, Johnson & Johnson disclosed the latest analysis results of the phase 2 DAHLIAS study on the investigational drug nipocalimab in adult patients with moderate to severe Sjögren's syndrome (SjD) at the American College of Rheumatology (ACR) meeting, showingKey indicators of disease activity improved in SjD patients receiving nipocalimab, with IgG significantly reduced by more than 77%.
Nipocalimab Is a potential best-in-class antibody therapy targeting FcRn;Is also expected to become the first for this target used in treatmentSjD'sMedicine.
About the latest research data
DAHLIAS is a Phase II multicenter, randomized, placebo-controlled double-blind study. The participants are adult patients with moderate to severe active primary SjD who are seropositive for anti-Ro60 and/or anti-Ro52 IgG antibodies. They are randomly assigned in a 1:1:1 ratio to receive intravenous nipocalimab (5 or 15 mg/kg) every two weeks or a placebo until Week 22, along with protocol-permitted background standard treatment. Safety assessments continue until Week 30.Results show:
At 24 weeks, patients receiving nipocalimab showed significant improvement in ClinESSDAIa scores, reaching the primary endpoint. Additionally, key secondary endpoints included multi-organ assessment (DALc), physician global assessment (PhGAd), and the Clinical Trial Endpoint Composite Tool (STARe, CRESSf); important SjD symptoms such as dry mouth, dry eyes, and vaginal dryness showed a trend of improvement.
Specifically, patients receiving 15 mg/kg treatment every two weeks showed a significant reduction in IgG levels (including autoantibodies); subjects with the highest baseline levels of anti-Ro and anti-La autoantibodies typically exhibited the greatest improvement in ClinESSDAI, which is associated with the substantial reduction in IgG and total IgG autoantibodies induced by nipocalimab. Additionally, at week 24, patients in the high-dose nipocalimab group (15 mg/kg) demonstrated improved objective salivary flow (i.e., an increase of at least 50% from baseline) compared to the placebo group (32.7% vs. 16%).
About Nipocalimab
Nipocalimab is designed to bind with high affinity to block FcRn and reduce levels of circulating immunoglobulin G (IgG) antibodies without affecting other immune functions.
In August 2020, Johnson & Johnson acquired Momenta, a biotechnology company, for $6.5 billion, gaining access to the Phase 3 drug nipocalimab and strengthening its pipeline in autoimmune diseases.
Currently, the drug has been granted by the FDA and EMA for Hemolytic Disease of the Fetus and Newborn (HDFN) and Warm Autoimmune Hemolytic Anemia (wAIHA),Generalized Myasthenia Gravis (gMG), Fast Track Designation for Fetal and Neonatal Alloimmune Thrombocytopenia (FNAIT);Granted FDA and EMA wAIHA, HDDN,Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)and FNAIT Orphan Drug Designation;Granted FDA's hddn Breakthrough Therapy;Granted hddn Orphan Drug Designation by EMA.
About Sjögren's Syndrome
Sjögren's syndrome is one of the most common autoantibody-driven diseases, and currently, there are no approved systemic therapies for its treatment. Clinically, it often presents with an insidious onset, commonly involving damage to the salivary and lacrimal gland functions, with serological characteristics marked by the significant production of autoantibodies and hyperimmunoglobulinemia. Over 50% of patients with Sjögren's syndrome suffer from moderate to severe disease, and the disease burden may be as high as that of rheumatoid arthritis or systemic lupus erythematosus, often leading to a reduced quality of life, immune dysfunction, and an increased risk of mortality.
Statistics show that about 4 million people worldwide are affected, with the incidence rate in women being 9 times that of men.
About FcRn Drugs
Part.1
Target Introduction
FcRn, also known as the Brambell receptor, is an atypical Fcγ receptor (FcγRs) encoded by the Fcgrt gene. The recognition of immunoglobulin (IgG) by FcγRs represents an important mode of immune regulation, which can bind to the conserved Fc domain of IgG and deliver unique antigenic determinants in the form of IgG immune complexes (IgG IC). These antigenic determinants are loaded onto MHC class I and II molecules, subsequently stimulating relevant CD8+ and CD4+ T cells. IgG is primarily synthesized and secreted by plasma cells in the spleen and lymph nodes, usually existing in monomeric form, and is the main antibody component in serum. In most humoral immune processes, the effector functions mediated by IgG are involved in protective actions against both viral and cellular pathogens. IgG can eliminate exogenous substances invading the human body, while FcRn is used in the human body to maintain IgG levels, extend the half-life of IgG antibodies, and slow down the degradation of IgG in lysosomes.
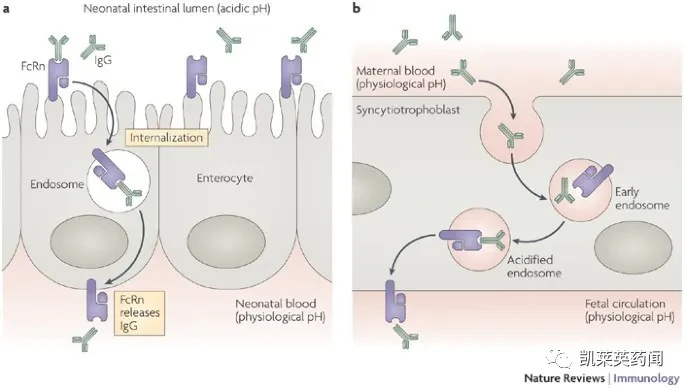
Studies have shown that FcRn is widely expressed in cells across various tissues, including endothelial cells aligned with blood vessel walls, intestinal epithelial cells, airway epithelial cells, placental syncytiotrophoblasts, hepatocytes, endothelial cells, myeloid cells, and bronchial duct cells, among others. Additionally, high levels of FcRn expression have been found in hematopoietic cells such as B cells, macrophages, and dendritic cells, which theoretically could lead to seventy or eighty types of autoimmune diseases. The antigen presentation process mediated by FcRn does not affect homeostatic immune activation in various locations but is an essential component of endogenous anti-tumor activity and the tumor immune surveillance system. This process is consistent with the widely recognized importance of CD8+ T cells in anti-tumor functions, thus FcRn-mediated cross-presentation can also initiate mechanisms that protect against tumors.
Part.2
Transaction Status
According to incomplete statistics, there have been nearly 9 deals related to FcRn. Apart from Johnson & Johnson's acquisition of Momenta's nipocalimab, other transactions mainly focus on drugs such as batoclimab, efgartigimod, and tifalibep.

Part.3
Progress of Drugs in Research
According to incomplete statistics, there are currently about a dozen FcRn drugs under research and development globally, mainly monoclonal antibodies; the approved indications mainly include myasthenia gravis, chronic inflammatory demyelinating polyneuropathy, and primary immune thrombocytopenia, etc.
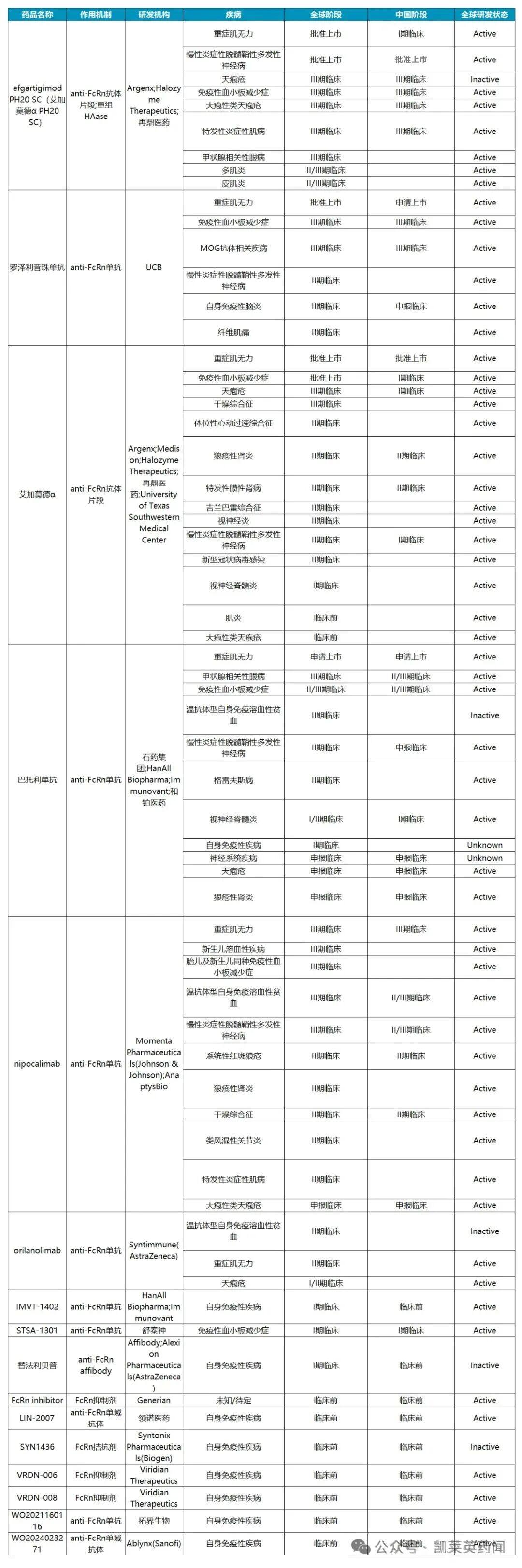
1. Efgartigimod
Efgartigimod is a novel therapeutic drug that is highly targeted to IgG for antibody-mediated autoimmune diseases and is the world's first FcRn inhibitor. Efgartigimod was approved by the FDA in December 2021 for the treatment of adult gMG who are positive for acetylcholine receptor (AChR) antibodies. In June 2024, the FDA approved the subcutaneous injection of Efgartigimod (VYVGART Hytrulo) for the treatment of CIDP.
Zai Lab acquired Efgartigimod from Argenx in January 2021 and holds the exclusive rights for development and commercialization in Greater China (including mainland China, Hong Kong, Macao, and Taiwan). In July 2023, the NMPA approved the biologics license application (BLA) for Efgartigimod Alfa Injection for use in combination with standard therapy to treat adult gMG patients who are AChR antibody positive. In the same month, the BLA for the subcutaneous injection formulation of the drug was accepted by the NMPA for the treatment of adult gMG. In November 2024, the NMPA approved the marketing application for the subcutaneous injection formulation of Efgartigimod for the treatment of adult CIDP patients, making it the first and currently the only drug approved in China for the CIDP indication.
In addition, Japan's Ministry of Health, Labour and Welfare (MHLW) approved the intravenous formulation of efgartigimod for the treatment of adult patients with primary immune thrombocytopenia (ITP) in March 2024. In the same month, the company announced that it would plan to advance a Phase 3 clinical study of efgartigimod in adult patients with SjD based on data analysis from the Phase 2 clinical study RHO.
gMG:In the pivotal Phase III ADAPT clinical trial, patients were randomized 1:1 to receive either efgartigimod or placebo. The results showed that the study met its primary endpoint: in acetylcholine receptor antibody-positive (AChR Ab+) gMG patients, a significantly higher proportion of patients in the efgartigimod group were responders compared to the placebo group (67.7% vs 29.7%; p<0.0001) based on Myasthenia Gravis Activities of Daily Living (MG-ADL) scores. Responders were defined as having at least a 2-point improvement on the MG-ADL score for four consecutive weeks or longer. Additionally, 40% of patients in the efgartigimod group achieved minimal manifestation status (defined as an MG-ADL score of 0 [asymptomatic] or 1), compared to only 11.1% in the placebo group. Among AChR-Ab+ responders, 84.1% of patients experienced clinically meaningful improvement in MG-ADL scores within the first two weeks of treatment. In terms of safety, the efgartigimod group was comparable to the placebo group (AEs: 77% vs 84%), with the most common adverse reactions being headache (29% vs 28%) and nasopharyngitis (12% vs 18%).
In addition, the Phase 3 ADAPT-SC study showed that, in adult gMG patients, the subcutaneous formulation was non-inferior to intravenous infusion in reducing total IgG on Day 29. On Day 29, compared with baseline levels, the mean reduction in total IgG was 66.4% for the efgartigimod alfa subcutaneous formulation and 62.2% for the intravenous formulation. This supports the marketing application for the subcutaneous formulation.CIDP:ADHERE Study is the largest clinical study to date for the treatment of CIDP; 69% (221/322) of patients receiving subcutaneous efgartigimod, regardless of prior treatment status, demonstrated clinical improvement, including improvements in mobility, function, and strength. The study met its primary endpoint (p<0.0001), showing that subcutaneous efgartigimod reduced the risk of relapse by 61% compared with placebo (HR: 0.39, 95% CI: 0.25, 0.61). The safety profile was consistent with previous findings from intravenous efgartigimod clinical studies and real-world use. Subgroup analysis of Chinese patients showed a 69% reduction in relapse rate with subcutaneous efgartigimod compared to placebo. Additionally, among Chinese patients treated in the open-label portion of the study, 78% demonstrated evidence of clinical improvement (ECI).
ITP:Results of the Global Phase 3 Clinical Study ADVANCE-IV Published in the September 2023 Issue of The Lancet Show that the Study Met its Primary Endpoint: A Higher Proportion of Chronic ITP Patients Treated with Efgartigimod Achieved Sustained Platelet Count Responses Compared to Placebo. The study demonstrated rapid efficacy of efgartigimod in treating both chronic and persistent ITP patients, with a 51% response rate based on the International Working Group (IWG) score (the IWG score is an assessment tool for primary immune thrombocytopenia developed by the International Working Group for ITP, highly relevant to clinical treatment). Responders to the primary endpoint were observed across different patient types, regardless of age, disease severity, time since diagnosis, prior ITP treatments, or concomitant medications. In this 24-week study, efgartigimod was well-tolerated, with safety and tolerability consistent with previous clinical studies.
2、Rozanolixizumab
Rozanolixizumab is a subcutaneously injectable humanized monoclonal antibody developed by UCB, which specifically binds to FcRn with high affinity; by blocking the interaction between FcRn and immunoglobulin G (IgG), it accelerates antibody catabolism and reduces the concentration of pathogenic IgG autoantibodies. In June 2023, the FDA approved the marketing application for Rystiggo (rozanolixizumab) for the treatment of adult patients with gMG.
In May 2023, the company released data from the Phase 3 MycarinG trial, a randomized, double-blind, placebo-controlled, adaptive two-stage design study. A total of 300 patients were randomly assigned to receive rozanolixizumab at doses of 7 mg/kg or 10 mg/kg, or placebo, administered once weekly via subcutaneous injection for a treatment period of 6 weeks, followed by an 8-week follow-up observation period. The primary endpoint was MG-ADL, and secondary endpoints included MGC, QMG, patient-reported outcomes (PRO), and MG-ADL; safety endpoints were treatment-emergent adverse events (TEAEs) leading to treatment discontinuation. The results showed:
(1) On Day 43 post-treatment, both the high- and low-dose groups showed significant improvement in MG-ADL, with changes from baseline of –3.40 and -3.37, respectively, compared to -0.78 in the placebo group; the drug treatment groups were significantly better than the placebo group. (2) Subgroup analysis of MuSK antibody-positive gMG patients showed an improvement of –4.16 in the high-dose group and -7.28 in the low-dose group, compared to 2.28 in the placebo group. (3) On Day 43 post-treatment, both the high- and low-dose groups showed significantly greater improvements in MGC, QMG scores, and PRO compared to the placebo group; symptom improvement was first observed on Day 8 post-treatment and persisted throughout the treatment period until returning to baseline on Day 99. (4) In the MuSK antibody-positive gMG subgroup, all patients receiving either the high or low dose experienced significant MG-ADL improvement (improvement ≥2 points), whereas only 14% (1/7) of the placebo group reached this level of improvement; regarding the percentage of patients achieving minimal symptoms post-treatment, the high-dose group was 28%, the low-dose group was 26%, and the placebo group was 3%. (5) A significant decrease in total IgG levels occurred on Day 8 post-treatment, then returned to baseline on Day 99.
In terms of safety, the incidence of any TEAEs or drug-related TEAEs was very low and similar to that in the placebo group; the incidence of treatment discontinuation due to TEAEs was slightly higher in the high-dose group, with symptoms including diarrhea, upper abdominal pain, vomiting, oral herpes, metastatic squamous cell carcinoma, pruritus, and deep vein thrombosis. No deaths, serious hypersensitivity reactions, or patients with suicidal tendencies occurred during the study period, and patients showed good tolerance to subcutaneous injection of the drug.
3. Batoclimab
Batoclimab (HBM9161) is an anti-FcRn monoclonal antibody that blocks the binding of FcRn-IgG, accelerating the clearance of IgG (including pathogenic IgG) in the body. It holds promise to bring a new generation of therapies for pathogenic IgG-mediated autoimmune diseases, including myasthenia gravis. In October 2022, CSPC Pharmaceutical Group reached a licensing agreement with Harbour BioMed, obtaining the rights for the development, production, and commercialization of Batoclimab in Greater China.
In March 2024, the company announced the results of a Phase III clinical study. This randomized, double-blind, placebo-controlled parallel study enrolled 132 adult patients with generalized myasthenia gravis. These patients were randomly assigned to either the batoclimab treatment group or the placebo group and received subcutaneous injections in 6-week cycles. The improvement in disease symptoms was assessed based on changes in the Myasthenia Gravis Activities of Daily Living (MG-ADL) score from baseline. Data showed that as early as the second week of treatment, the MG-ADL score improvement curve for the batoclimab group significantly diverged from the control group, indicating rapid efficacy and symptom relief. By Day 43, after completing the first treatment cycle, the sustained improvement rate (proportion of patients with an ADL score improving by 3 points from baseline and maintained for 4 consecutive weeks) in the batoclimab group reached 58.2%, compared to 31.1% in the control group. Additionally, within the first treatment cycle, 25.4% of patients in the batoclimab group achieved minimal symptom expression (MSE, defined as an ADL score of 0 or 1), far exceeding the 4.7% in the control group. Moreover, similar trends favoring the batoclimab group over the control group were observed in the Quantitative Myasthenia Gravis (QMG) score, Myasthenia Gravis Composite (MGC) score, and the 15-item Myasthenia Gravis Quality of Life (MG-QOL15) assessment. In terms of safety, the incidence of treatment-emergent adverse events (TEAEs) was comparable between the batoclimab and control groups, with overall good tolerability and safety profiles.
4、Orilanolimab
Orilanolimab (SYNT001) is an FcRn monoclonal antibody that has the potential to improve the treatment of various rare diseases mediated by IgG. The drug was initially developed by Syntimmune and later acquired by Alexion. In December 2020, AstraZeneca proposed to acquire Alexion for approximately $39 billion, bringing the drug into AstraZeneca's pipeline. Currently, orilanolimab is being evaluated for the treatment of patients with warm autoimmune hemolytic anemia (WAIHA) and patients with pemphigus vulgaris (PV) or pemphigus foliaceus (PF).
References
1. Official Websites of Various Companies
2. CITIC Securities, Southwest Securities




"Views"Click once