Daiichi Sankyo's FLT3 Inhibitor Approved in China; Hengrui's Class 1 Drug Gains Third Indication; Domestic Long-Acting GLP-1 Triple Agonist Enters Phase 3 — Insight Weekly Drug Pipeline Report

GSK
Pharmaceutical R&D Manufacturer

Spero Therapeutics
Antibiotic Therapy Developer

According to the "Global New Drugs" module of the Insight database, this week (June 14–June(May 20) Globally, the development progress of 59 innovative drugs (including improved new drugs) advanced to a new stage, among which 2 were approved for marketing for the first time, 1 was submitted for marketing approval for the first time, and 5 entered Phase III clinical trials for the first time., 14 products were registered for the first time in Phase I clinical trials.
Below, Insight will highlight key project updates from China and abroad this week.
*Data Note: The data collection time for this weekly report is 2026-6-21 14:45. Due to the continuous high-speed updates of the Insight database, there may be timeliness differences in search results at different times. Please refer to the latest query.

On June 17 local time, GSK and Spero Therapeutics jointly announced that the U.S. Food and Drug Administration(FDA)Approved its oral antibiotic Utebzi (tebipenem pivoxil) For the treatment of complicated urinary tract infections in adults(cUTIs), including pyelonephritis, these infections are caused by certain susceptible pathogens, and patients have limited or no oral treatment options available.
This isThe First and OnlyApproved for use in such patientsOral Carbapenem Antibiotics。
This approval was supported by the Phase III PIVOT-PO trial(NCT06059846)Support for Positive Results.The PIVOT-PO trial is a global, randomized, double-blind, pivotal, non-inferiority(Non-inferiority margin: -10%)Phase III trial designed to evaluate the therapeutic potential of oral Utebzi compared with intravenous imipenem-cilastatin in hospitalized adult patients with complicated urinary tract infections, including acute pyelonephritis.
This trial demonstrates that in the treatment of complicated urinary tract infections(including pyelonephritis)of hospitalized patients, based on the overall response at the test-of-cure visit(Composite Endpoint of Clinical Cure and Microbiological Eradication), Utebzi demonstrated non-inferiority compared to intravenous imipenem-cilastatin.Utebzi(Oral, 600 mg)The overall success rate was 58.5%.(261/446), andImipenem-Cilastatin(Intravenous injection, 500 mg)The overall success rate was 60.2%.(291/483) (Adjusted treatment difference: -1.3%; 95% CI: -7.5%, 4.8%)。
The safety profile of Utebzi is generally similar to that of imipenem-cilastatin and other carbapenem antibiotics. The most commonly reported adverse events(Incidence ≥3%)for diarrhea and headache; these events were all mild or moderate in severity and not serious.
Carbapenems are the standard of care for treating severe or resistant infections, but to date, they can only be administered intravenously, which increases the utilization of hospital resources and reduces patients’ quality of life. Following its approval, Utebzi is expected to become an effective oral alternative that can be used outside the hospital setting.
In September 2022, GSK and Spero entered into an exclusive licensing agreement, granting GSK the rights to develop and commercialize the drug in all markets except for certain regions in Asia.
On June 19 local time, Sanofi announced that Japan’s Ministry of Health, Labour and Welfare has approved Sarclisa(Isatuximab)Subcutaneous Injection(SC)Dosage Form in Combination with Approved Standard Regimens for the Treatment of Multiple Myeloma(MM)。
The indications approved in Japan for the subcutaneous formulation of Sarclisa include:
With Pomalidomide and Dexamethasone(Pd)in combination with, or in combination with carfilzomib for the treatment of relapsed or refractory multiple myeloma(R/R MM),
with bortezomib, lenalidomide, and dexamethasone(VRd)Used in Combination for the Treatment of Newly Diagnosed Multiple Myeloma(NDMM)Adult Patients.
Additionally, the CirCLIQ on-body injector based on the enFuse platform submitted by Enable Injections(OBI)The regulatory application is under review in Japan., which is an auto-injector device for the subcutaneous administration of Sarclisa. If approved, the subcutaneous formulation of Sarclisa may becomeThe First Anti-Cancer Therapy Administered via a Portable Injector, it will also become the first multiple myeloma drug in Japan to offer both manual subcutaneous injection and administration via an on-body injector.
This approval is based on the Phase 3 clinical study IRAKLIA(NCT05405166)Positive Outcomes, this study demonstrated that the subcutaneous formulation is comparable in efficacy to the intravenous formulation, with additional supportive studies available. In addition to manual subcutaneous injection, these studies also evaluated administration via wearable injection devices(OBI)Sarclisa subcutaneous injection formulation for administration.
In the IRAKLIA study, for adult patients with relapsed or refractory multiple myeloma who have received at least one prior line of therapy, via a portable injection deviceSubcutaneous Injection of Sarclisa in Combination with Pomalidomide and Dexamethasone(Pd)TherapeuticObjective Response Rate(ORR)71.1%, whileIntravenous Sarclisa in Combination with PdTherapeuticObjective Response Rate was 70.5%, demonstrating that the efficacy of the subcutaneous formulation is non-inferior to that of the intravenous formulation(Hazard ratio: 1.008; 95% confidence interval: 0.903 - 1.126; p = 0.0006)。
The overall safety profile of subcutaneous Sarclisa in combination with Pd therapy observed in this study was consistent with the established safety profile of intravenous Sarclisa in combination with Pd therapy. Infusion reactions occurred in 25% of patients receiving intravenous Sarclisa plus Pd, compared with 1.5% of patients receiving subcutaneous Sarclisa plus Pd.
except for injection via portable injection devices in 0.4% of cases(n=19/5,145 injections)mild local injection site reactions observed in(ISR)No new safety issues were observed. Nearly all injection site reactions were Grade 1, with only one case being Grade 2.
Multiple myeloma is the third most common hematologic malignancy in Japan.In Japan, the intravenous formulation of Sarclisa is currently approved for five indications., including in combination with VRd for newly diagnosed multiple myeloma(NDMM), and for relapsed/refractory multiple myeloma(R/R MM)Four Different Treatment Regimens(in combination with Pd, in combination with carfilzomib and dexamethasone (Kd), in combination with dexamethasone alone, or as monotherapy)。
June 8, 2026: Sarclisa Subcutaneous Injection Administered via the CirCLIQ OBI Device and Manual InjectionAlready approved in the EU for the treatment of MM patients, covering all approved indications and combination regimens for the intravenous formulation of Sarclisa. In addition, the application for the subcutaneous formulation of Sarclisa is currently under review in both the United States and China.
Source: Corporate Website
The acceptance of this sBLA is based on the results of the Phase III SUNMO study. At a median follow-up of 23.2 months, compared withRituximab, Gemcitabine and Oxaliplatin(R-GemOx)Compared with the regimen, the Lunsumio VELO combined with Polivy regimen reduces the risk of disease progression or deathReduced by 59%(HR 0.41,p<0.0001), andMedian PFS Extended to 11.5 Months, is the R-GemOx regimen(3.8 months)three times.
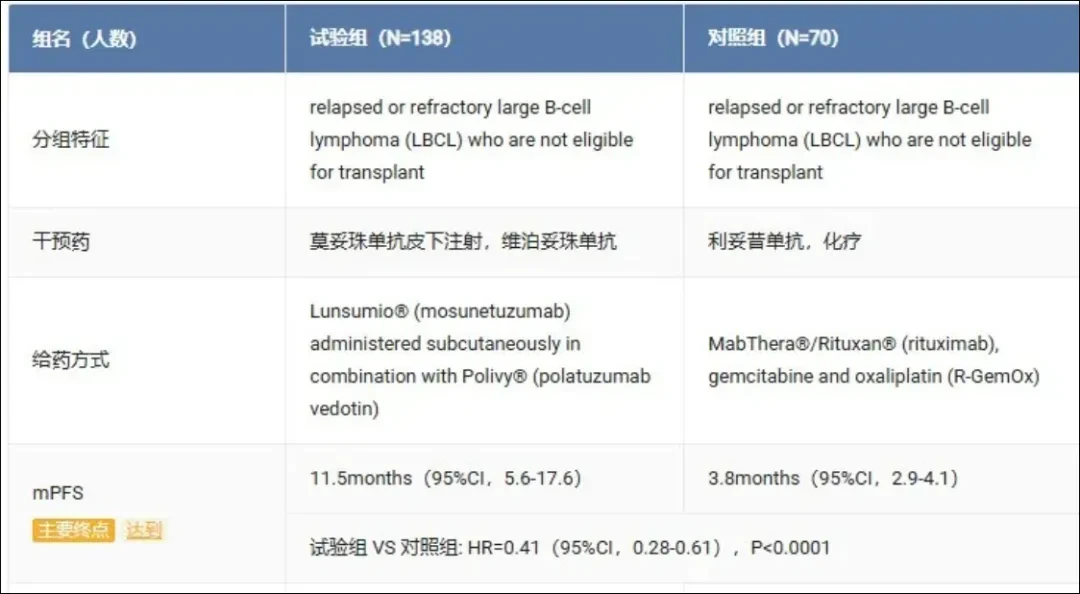
The safety profile of the Lunsumio and Polivy combination regimen is consistent with the known safety profiles of the individual study drugs. Cytokine release syndrome in the Lunsumio VELO plus Polivy treatment group(CRS)ofLow incidence, accounting for only one-quarter, with fewer than 5% of patients experiencing Grade 2 or 3 CRS.
Updated data from this trial were recently presented at the ASCO and EHA conferences, showing that with extended follow-up, this combination regimen demonstrated improved progression-free survival(PFS)AspectContinued demonstration of clinical benefit, especially inSecond-line treatmentand no new safety signals were identified.
Lunsumio(Mosunetuzumab)is a type ofFirst-in-class CD20 x CD3T cell-engaging bispecific antibodies have been approved for the treatment of third-line or later relapsed or refractoryFollicular Lymphoma (FL) Adult patients, with intravenous and subcutaneous injections(Lunsumio VELO™)Two dosage forms.
Polivy(Polatuzumab Vedotin)is a type ofFirst-in-Class CD79b ADC, which has currently received widespread global approval, can be used in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone for the treatment of previously untreated patients(First-line)of DLBCL, and can also be used in combination with bendamustine and rituximab for the treatment of relapsed or refractory DLBCL. In 2025, the drug's annual sales reached$1.777 billion。
On June 15 local time, AstraZeneca announced that Alexion, its rare disease division, had submittedUltomiris(Ravulizumab)Supplement to Biologics License Application(sBLA)Accepted by the U.S. FDA and granted Priority Review designation for the treatment of adult IgA nephropathy(IgAN). Its PDUFA decision date is expected to beQ4 2026。

Source: Corporate Official Website
This sBLA is based on the pre-specified interim analysis results from the Phase III I CAN trial, which were recently presented at the 2026 European Renal Association(ERA)Announced at the conference.
I CAN(ALXN1210-IgAN-320)This is a global, Phase III, randomized, double-blind, placebo-controlled trial designed to evaluate the efficacy and safety of Ultomiris in adult patients with immunoglobulin A nephropathy (IgAN) at risk of disease progression. Prior to screening, participants must have received stable concomitant IgAN therapy consistent with standard of care for at least three months.
In this trial, 5.6% of patients receiving placebo(95% confidence interval [CI]: -4.9%, 15.0%)In comparison,Ultomiris reduced the 24-hour urine protein-to-creatinine ratio at Week 34(UPCR)A 46.6% reduction from baseline(95% confidence interval: 39.0%, 53.2%), the placebo-adjusted treatment effect was 43.4%(95% confidence interval: 33.5%, 51.8%; p < 0.0001)。
Rapid reduction in proteinuria was observed as early as Week 10 with Ultomiris(36.7% [95% CI:30.2%,42.6%]), and continued until week 34, whereas the placebo group was 8.5%.(95% CI:0.5%,15.8%). These results were consistent across patient subgroups, reflecting diverse demographic and baseline clinical characteristics as well as disease severity. The primary endpoint of the trial—estimated glomerular filtration rate(eGFR)Changes from baseline will be measured at Week 106.
The safety profile observed in the I CAN trial was consistent with the known safety profile of Ultomiris, with overall good tolerability and no new safety concerns identified.
Pharmaceutical Transactions
According to the Insight database, a total of 15 transaction events occurred this week (June 14–June 20).
On June 16 local time, neuroscience company 4E Therapeutics, Inc. announced that it had been acquired by Eli Lilly and Company.
4E is committed to developingNext-Generation Non-Opioid Therapies for Chronic Pain, currently developing a series of oral MNK inhibitors aimed at treating chronic pain by targeting the MNK-eIF4E signaling pathway in peripheral sensory neurons. These compounds are designed to provide significant pain relief while avoiding the central nervous system effects associated with many existing therapies.
4ET1103 It is the company’s first MNK inhibitor to enter human clinical trials for the treatment of pain, and has demonstrated a favorable safety profile in Phase 1 studies.
Screenshot from: GSK official website
2、Everest Medicines Enters Exclusive Commercial License Agreement to In-License Novel Kidney Disease Drug
On June 16, Everest Medicines announced that it had entered into an exclusive commercialization license agreement with Dimerix Limited, obtaining the rights to DMX-200 in Greater China(including Mainland China, Hong Kong SAR, Macao SAR, and Taiwan region of China), South Korea and several Southeast Asian countries(including Singapore, Malaysia, Thailand, Indonesia, Vietnam, and the Philippines)the rights to clinical development and commercialization.
This collaboration will further optimize Everest Medicines’ nephrology product portfolio, enhance pipeline synergy efficiency, and strengthen the company’s strategic position in the fields of kidney diseases and autoimmune disorders.
Pursuant to the agreement, Everest Medicines will pay Dimerix$10 million upfront payment, andUp to $30 million in development and regulatory milestone paymentsandCommercialization milestone payments of up to $300 million. In addition, Dimerix will receive royalties based on the future annual net sales of DMX-200 in the licensed territory.10% to 15% Tiered Royalties。
DMX-200 is a chemokine receptor 2(CCR2)small-molecule inhibitor currently under development for the treatment of focal segmental glomerulosclerosis(FSGS)the global pivotal Phase III clinical study ACTION3. The study has completed the enrollment of 333 patients worldwide. Positive results from the interim analysis announced in early 2024 showed that DMX-200 was significantly superior to placebo in reducing proteinuria.
DMX-200 has been granted orphan drug designation by the U.S. FDA and the European EMA.

Source: Official Company
3BioMap and Harbour BioMed Announce Major Strategic Partnership
June 15,BioMap(BioMap)andHarbour BioMed(Stock Code: 02142.HK)Jointly announced that the two parties will establish a multi-level, long-term comprehensive strategic partnership,Comprehensive Collaboration on AI-Driven R&D of Complex Macromolecular Drugs, aiming to systematically address the bottlenecks and challenges in the research and development of next-generation innovative therapies, and jointly build a globally competitive R&D ecosystem.
Under the strategic cooperation framework, the two parties will jointly establish a new AI-driven pipeline R&D company targeting the global market—MegaStream TechBio.The company will integrate its “exclusive big data × proprietary large models × extensive innovative pipeline portfolio” to build an AI R&D engine centered on a next-generation intelligent wet-and-dry closed-loop experimental platform and personalized, multimodal, multi-attribute generative large models. Its focus is on addressing major unmet clinical needs in cardiovascular disease, renal disorders, anti-aging, and oncology, with a strategy centered on First-in-Class(FIC)and Best-in-Class(BIC)With this as the core benchmark, we are developing next-generation, globally competitive, cutting-edge complex macromolecular drugs, systematically advancing our differentiated innovative pipeline into clinical stages, and striving to become a global leader and benchmark enterprise in AI-driven R&D of complex macromolecular therapeutics.
Harbour BioMed, as the lead initiator, will open up its globally exclusive fully human antibody platform, extensive target biology expertise, and global clinical development capabilities to MegaStream; BioMap, as a co-initiator, will provide foundational AI technology enablement, model engineering support, and intelligent R&D capabilities to help accelerate the company’s pipeline development.
MegaStream’s initial pipeline comprises AI-driven drug discovery projects from the parties’ prior collaborations alongside newly initiated AI-native pipeline development projects. In accordance with industry standards, the initiating parties will be entitled to potential upfront payments, success-based milestones, and royalty shares. Meanwhile, MegaStream’s core management team will be jointly formed by senior executives from multinational pharmaceutical companies and leading experts in the field of artificial intelligence, with relevant personnel currently being onboarded.
Source: Company Announcement


This was a randomized, double-blind, placebo-controlled study that enrolled 539 patients with newly diagnosed FLT3-ITD–positive acute myeloid leukemia (AML). Patients were randomly assigned in a 1:1 ratio to receive quizartinib or placebo in combination with standard induction and consolidation therapy, followed by monotherapy maintenance treatment.
The results showed that,The mOS in the quazatinib group was31.9 months(95%CI,21.0-NE), while the placebo group was 15.1 months(95%CI,13.2-26.2), HR was 0.776(95% CI: 0.615-0.979;P=0.0324)。
In addition, Daiichi Sankyo has also registered another item in China.Phase III StudyQuANTUM-Wild(NCT06578247/CTR20244780),This is aAn international, multicenter, double-blind, randomized, placebo-controlled clinical trial aimed atEvaluation of Quinazetinib or Placebo in Combination with Induction and Consolidation Chemotherapy, and Quinazetinib or Placebo Monotherapy as Maintenance TreatmentNewly Diagnosed FLT3-ITD-Negative AML Patientsefficacy and safety. The Insight database shows that the study plans to enroll 700 patients globally, including 105 in China. The first subject was dosed in China in April 2025.
2. BMS: New Indication for Oral TYK2 Allosteric Inhibitor Approved for Launch in China

Deucravacitinib(Deucravacitinib)is a tyrosine kinase 2(TYK2)Inhibitor,By selectively targeting TYK2 to inhibit the signaling of IL-23, IL-12, and IFN, which are key cytokines involved in the pathogenesis of multiple immune-mediated diseases.September 2022,Deucravacitinib Approved for Marketing in the United States(Brand Name:Sotyktu), for the treatment of plaque psoriasis in adults, becomingWorld's FirstApproved TYK2 Allosteric Inhibitor.
In China,DeucravacitinibOctober 2023First approved for marketing, indicated for the treatment ofAdult patients with moderate-to-severe plaque psoriasis eligible for systemic therapy or phototherapy; and its approval for psoriatic arthritis marks the second indication approved for this drug in China.
Among them, POETYK PsA-1 enrolled approximately 670 patients with active psoriatic arthritis (PsA) who had not previously receivedbDMARD treatmenttherapy. POETYK PsA-2 enrolled approximately 730 patients with active psoriatic arthritis (PsA) who were either naive to biologic disease-modifying antirheumatic drugs (bDMARDs) or had previously received tumor necrosis factor-alpha (TNFα) inhibitor therapy. The primary endpoint for both studies was achieving the American College of Rheumatology 20% improvement criteria (ACR20) at Week 16.(ACR20,≥20% improvement in disease signs and symptoms)Proportion of Participants with Remission.
Both trials met their primary endpoints.POETYK PsA-1 data show that,At Week 16,AcceptDeucravacitinibThe proportion of treated patients achieving ACR20 response was significantly higher, respectively 54.2% vs 34.1%(p < 0.0001)。
Meanwhile, the trial met multiple key secondary endpoints.At Week 16,Patients treated with deucravatinib showed improvements in a range of clinical measures of disease activity, patient-reported outcomes, and extra-articular manifestations of psoriatic arthritis, including the Psoriasis Area and Severity Index.(PASI)75% improvement in response rate and Health Assessment Questionnaire Disability Index(HAQ-DI)Score, 36-Item Short Form Health Survey(SF-36)Total Score of Physical Health(PCS)and minimal disease activity(MDA)Response rate. Furthermore, the response rates for ACR50 and ACR70 also improved.
No new safety signals were identified in the POETYK PsA-1 trial. The most common adverse events in the deucravacitinib and placebo groups(AE)All are upper respiratory tract infections(5.1% and 3.0%, respectively)。
The POETYK PsA-2 study results showed that,At Week 16, the efficacy of deucravacitinib was superior toIn the placebo group, through Week 52, patients who continued or switched to deucravacitinib showed sustained improvement in clinical response, and efficacy was maintained in those who continued treatment with deucravacitinib.
Specifically,
At Week 16, among patients receiving deucravacitinib treatment54.2% Achieved ACR20, while the placebo group was 39.4%(p = 0.0002);
At Week 52, among patients who continued to receive deucravacitinib treatment62.2% achieved ACR20 response criteria, 67.3% of patients who switched from placebo to deucravacitinib after Week 16 achieved an ACR20 response. InACR50 and ACR70A similar trend was also observed in terms of palliative care standards;
Compared with the placebo group, at Week 52, the deucravacitinib groupKey Secondary EndpointsSustained maintenance of favorable outcomes, including PASI75 response, proportion achieving MDA, HAQ-DI score, and SF-36 PCS score;
Through Week 52, deucravacitinibWell Tolerated, and its safety profile was consistent with previous findings in psoriatic arthritis and psoriasis.
According to the Insight database, in addition toPlaque Psoriasis andexcept for active psoriatic arthritis,Deucravacitinib is also being developed forSjögren's Syndrome(Clinical Phase III)Lupus Erythematosus(Clinical Phase III). Crohn's disease(Phase II Clinical Trial)and other indications.
This approval is based onA Multicenter, Randomized, Double-Blind, Phase III Clinical Trial of Dalpiciclib Combined with Endocrine Therapy in the Adjuvant Treatment of HR-Positive, HER2-Negative Female Breast Cancer(Study Code: DAWNA-A)positive outcomes.
The DAWNA-A study enrolled over 5,000 participants, who were randomized in a 1:1 ratio to receive either dalpiciclib plus endocrine therapy or placebo plus endocrine therapy.The primary endpoint of the study was invasive disease-free survival in patients.(IDFS), defined as the time from randomization to the first occurrence of any of the following events: ipsilateral or contralateral breast cancer recurrence, regional or distant recurrence, or death from any cause.
At the 2025 ASCO Annual Meeting, Hengrui announcedDAWNA-A StudyResults of the Prespecified First Interim Analysis.As of the data cutoff date, the median follow-up time was 20.3 months.
Results showed that, compared with placebo combined with endocrine therapy(ET)In comparison, Dalpiciclib combined with ETSignificantly prolonged iDFS(HR 0.56, 95% CI 0.43–0.71; one-sided P<0.0001), and in2-year absolute iDFS benefit of 4.5%。
Furthermore, the iDFS benefit conferred by dalpiciclib was generally consistent across all stratification factors and other baseline subgroups. Disease-free survival(DFS)and distant disease-free survival(DDFS)It also demonstrated that dalpiciclib combined with ET was superior to placebo combined with ET. In terms of safety, the overall safety profile of dalpiciclib was manageable.
Dalpiciclib is an orally administered, selective small-molecule CDK4/6 inhibitor independently developed by Hengrui Medicine, and alsoChina's FirstNovel High-Selectivity CDK4/6 Inhibitor Independently Developed
Previously, the drug had already received NMPA approval for marketing in two indications: 1)Combination with Fulvestrant for the Treatment of Recurrent or Metastatic Breast Cancer That Is HR-Positive and HER2-Negative and Has Progressed After Endocrine Therapy;2)Combined aromatase inhibitors as initial therapy are indicated for patients with HR-positive, HER2-negative locally advanced or metastatic breast cancer. This marks the drug's approvalItem 3 Indication。

In June 2024, Zeltigen Pharmaceuticals announcedPhase III Pivotal Trial of Garsocitinib for the Treatment of Severe Alopecia AreataMet the primary efficacy endpoint。This is a multicenter, randomized, double-blind, placebo-controlled, parallel-group Phase III clinical trial.(Protocol No.: ZGJAK018), 425 patients with severe alopecia areata who met the protocol requirements were randomly assigned to receive ritlecitinib tablets 50 mg twice daily, 75 mg twice daily, or placebo tablets.
In 2025Annual Meeting of the European Academy of Dermatology and Venereology(EADV)on,Zelgen Pharmaceuticals announced the specific results of the trial. The primary efficacy endpoint data showed that after 24 weeks of treatment, the Severity of Alopecia Tool scale(SALT)Percentage of subjects with a score ≤20,Ruxolitinib Tablets in Both Groups Were Significantly Superior to the Placebo Group(50 mg:34.5%; 75 mg: 28.0%; placebo: 3.5%), achieving statistical significance。
In terms of safety, ruxolitinib demonstrated a favorable safety and tolerability profile in patients with severe alopecia areata.Most Common Adverse Events(occurring in ≥10% of patients and at a higher rate than in the placebo group)For decreased neutrophil or white blood cell counts, elevated creatine kinase or serum lactate dehydrogenase levels, increased platelet counts, upper respiratory tract infections, and hyperlipidemia. No deaths, malignancies, or thromboembolic events occurred.

As the second drug approved globally for the treatment of premature ejaculation,PSD502 With over 10 years of usage experience in Europe,Its efficacy and safety have been validated in clinical trials and real-world practice.. PSD502 treatment for 3 months, intravaginal ejaculatory latency time in patients with premature ejaculationIncreased from a baseline of 0.56 minutes to 2.6 minutes, significantly improving the scores for ejaculation control, sexual satisfaction, and distress in the various dimensions of the Premature Ejaculation Diagnostic Tool (PEDT).
In China,Plethora Solutions andFosun Pharma has issued a total ofThree Clinical Trials, two Phase I studies and one Phase III study.
Two Phase I Clinical Trialswere conducted separately in healthy Chinese males and females. The results showed that,Among subjects receiving PSD502, 38.9% of males and 66.7% of females experiencedAdverse Events Occurring During Treatment(TEAE), the incidence of TEAEs in both male and female subjects in the placebo group was 50%, all of which were mild to moderate and transient TEAEs. There were no serious adverse events or adverse events leading to study discontinuation. After multiple administrations, lidocaine and prilocaine in the subjects' bodiesQuick Clear, with a t1/2 of approximately 4 h, and Cmax values all lower than the systemic exposure levels expected to be associated with systemic toxicity(5000 ng/mL)1/27.
The ongoing Phase III study is aA multicenter, randomized, double-blind, placebo-controlled study designed toTo evaluate the efficacy, safety, and tolerability of PSD50 in Chinese subjects with premature ejaculation, a study enrolling 295 subjects was completed in February 2023. Specific study data have not yet been disclosed.
Mezigdomide is a specially optimizedNovelcerelon E3 LianLigaseModulator (CELMoD)Drugs, their myeloma cell killingImpairs immune activation capacity compared to traditional immunomodulators(IMiD)Further enhancement of the drug. Currently, clinical studies are being conducted for this product in the indications of multiple myeloma and acute myeloid leukemia.
March 2026, BMS announces mEzigdomide’s first Phase III SUCCESSOR-2 study yielded positive results in the interim analysis. The study aims toEvaluation of Mezigdomide in Combination with Carfilzomib and Dexamethasone(MeziKd)Versus Carfilzomib Combined with Dexamethasone(Kd)in Relapsed or Refractory Multiple Myeloma(RRMM)Efficacy and Safety in Patients。
The results showed that, compared with carfilzomib plus dexamethasone alone, oral mezigdomide in combination with carfilzomib andDexamethasoneDemonstrated statistically significant and clinically meaningful efficacy in the treatment of patients with relapsed or refractory multiple myelomaProgression-Free Survival(PFS)Improvement。
Safety is controllable andPredictable,The mortality rates for the MeziKd and Kd regimens were 21.5% and 26.7%, respectively, with disease progression being the primary cause of death.
This study is an international, multicenter, randomized, double-blind, active-controlled, non-inferiority Phase III clinical trial designed to compare the intravitreal injection of HLX04-O with that of ranibizumab.(IVT)Efficacy and Safety of Administration in Patients with wAMD. Enrolled patients were randomized in a 1:1 ratio to receive HLX04-O (1.25 mg) or ranibizumab (0.5 mg) IVT administration once every four weeks, continuing treatment for one year unless the patient dies, withdraws informed consent, is lost to follow-up, or the sponsor terminates the study.
The primary endpoint of this study wasBest Corrected Visual Acuity at Week 36(BCVA)Mean Change from Baseline, the key secondary endpoint was the mean change in BCVA from baseline at Week 48, and the other secondary endpoints included additional efficacy, safety, tolerability, and pharmacokinetic parameters.
The study results showed that,The mean changes from baseline in BCVA at Week 36 and Week 48 in the HLX04-O group were non-inferior to those in the ranibizumab group, meeting the primary and key secondary endpoints.. Furthermore, HLX04-O demonstrated a favorable safety profile, with overall, ocular, and non-ocular safety characteristics similar to those of ranibizumab in patients with wet age-related macular degeneration (wAMD).
LX04-O is Hanbeitai® independently developed by Henlius(Bevacizumab Injection)on the basis of optimizing the formulation, specifications, and manufacturing process of Hanbeitai® according to the requirements for ophthalmic medications, while keeping the active ingredient unchanged, developedNew Ophthalmic Formulation Products, for the treatment of wAMD.
In April 2025, HLX04-O met its primary endpoint in a Phase III clinical study conducted in Chinese patients with wet age-related macular degeneration (wAMD). In August 2025,Marketing Authorization Application for HLX04-O in the Treatment of wAMD (NDA) Accepted by the Center for Drug Evaluation of the National Medical Products Administration (NMPA).
Notably, Henlius previously licensed the exclusive global rights for ophthalmic therapeutic uses and/or treatments of Hanbeitai® to Essex, with an agreement to jointly develop the related products. Henlius and Essex will bear 20% and 80%, respectively, of the costs and expenses associated with the development activities for such products.
On June 18, local time,Nicox SA announced that its exclusive licensing partner in ChinaOcuvantisReceived positive pre-submission regulatory feedback from the CDE on NCX 470.Ocuvisionbelieves that this feedback is sufficient to support its submission of NCX 470 to the CDEMarketing AuthorizationApplication Documents.
NCX470(OT-301, gevaprost)is a novel compound combining a nitric oxide donor with prostaglandin synthesis, featuring a dual mechanism of action to lower intraocular pressure, intended forLowering intraocular pressure in patients with open-angle glaucoma or ocular hypertension。
December 2018,Ocuvision obtains exclusive license from Nicox toGreater ChinaDevelopment, manufacturing, contract manufacturing, import, export, use, distribution, marketing, promotion, offer for sale, and saleNCX470, and in March 2020, expanded the exclusive rights toSouth Korea and 12 countries in Southeast Asia。
In August 2025, Ocuvix and Nicox jointly announcedNCX470 ofThe Second International Multicenter Phase IIIClinical Research(Denali Study)The primary endpoint was met. Prior to this, the two parties had already collaborated to complete the drug'sThe First International Multicenter Phase IIIClinical Research(Mont Blanc Study), the results of the two studies will be jointly used to supportNCX470 New Drug Applications in China and the United States.
Nicox pointed out in its press release that itsKowa, the exclusive partner in the United States, will alsoThis SummerSubmit the New Drug Application for NCX 470 to the United States, followed shortly by an application submission to China.
On June 15, the Drug Clinical Trial Registration and Information Publicity Platform showed that The United Laboratories registered aUBT251 Injection Phase III (Overweight/Obesity) Clinical trial.
This is aA randomized, double-blind, parallel-group, placebo-controlled Phase III study designed toEvaluation of UBT251 Injection inOverweight/ObesityEfficacy and Safety in Trial Participants. The Primary Endpoint IsAdministration for 52 weeks,Percentage change in body weight from baseline andProportion of Trial Participants with a Weight Reduction of ≥5% from Baseline。
UBT251 is aLong-Acting GLP-1/GIP/GCG Triple Receptor AgonistTo date, clinical trials have been approved in China and/or the United States for multiple indications, including type 2 diabetes in adults, overweight/obesity, chronic kidney disease, and metabolic dysfunction-associated steatohepatitis (MASH).
In March 2025, Novo Nordisk $2 billionObtainUBT251 Global(excluding mainland China, the Hong Kong Special Administrative Region, the Macao Special Administrative Region, and Taiwan)The rights to develop, manufacture, and commercialize. Fed Pharma will retain the rights to UBT251 in Mainland China, the Hong Kong Special Administrative Region, the Macao Special Administrative Region, and Taiwan.
Furthermore, the proportion of participants in each UBT251 dose group who experienced a body weight reduction of ≥5% from baseline reached up to98.1%; The proportion of participants with a weight reduction of ≥10% from baseline reached up to 89.8%, and the proportion of participants with a weight reduction of ≥20% from baseline reached up to 48.4%.
On June 17, the Drug Clinical Trial Registration and Information Publicity Platform showed that,Subsidiary of Qingfeng PharmaceuticalSpero Therapeutics registered aDN022150 Monotherapy versus ChemotherapyAdvanced Pancreatic Cancer Harboring KRAS G12D MutationPhase III study.
DN022150 isDeveloped by Kerui PharmaceuticalOneSpeciesNovel Highly Selective KRAS G12D Inhibitor, by specifically binding to the Switch-II pocket of the mutant KRAS G12D protein, while also acting on the GDP-bound state of KRAS G12D(OFF)GTP-bound state(ON), inhibits KRAS-mediated signal transduction and downstream activation of the RAS-MAPK pathway, thereby exerting antitumor effects.
The Insight database shows that previouslyKerui Pharma has initiated two clinical trials for this drug, one of which isEvaluation of DN022150 in patients harboring KRAS G12D mutationsAdvanced Solid TumorsPhase I/IIa clinical trials in patients; another is to evaluateDN022150 CombinationGemcitabine/Albumin-bound Paclitaxel(AG)PlanFirst-line TreatmentCarrying KRASG12D gene mutationLocally Advanced or Metastatic Pancreatic CancerPhase I clinical trial.
Notably, the results of the Phase I/II clinical trials were presented as an oral report at the 2026 ASCO Annual Meeting.
As of April 5, among the 31 evaluable cases of pancreatic cancer(2L+)Among patients:Overall Objective Response Rate(ORR)41.9%、Disease Control Rate(DCR)93.5%。
DN022150 demonstrated a favorable overall safety and tolerability profile. Among all 67 patients evaluated for safety, 32.8%(22/67)of patients experienced ≥ Grade 3 treatment-related adverse events(TRAE)。InPhase II Recommended Dose(RP2D,650 mg)27.7% in the group(10/36)patientsGrade ≥3 TRAEs occurred. No grade 4–5 treatment-related adverse events were observed.
Based on the currently available clinical data, compared with the publicly reported clinical data of oral candidate drugs with the same target, DN022150 demonstrates advantages in gastrointestinal tolerability and the incidence of ≥ Grade 3 TRAEs.Competitive Safety Profile, holding promise to support long-term continuous treatment for patients.
This is the first Phase III clinical trial initiated for this drug.This is aA multicenter, open-label, randomized, controlled clinical trial designed toComparison of DN022150 for Injection versus Investigator-Selected Chemotherapy in Advanced Pancreatic Cancer Harboring KRAS G12D MutationsSecond-line and aboveefficacy. The trial plans to enroll 360 subjects in China, with the primary endpoint beingAccording to the Response Evaluation Criteria in Solid Tumors(RECIST1.1), by blinded independent central imaging assessment(BICR)ofProgression-Free Survival(PFS)and overall survival(OS)。
