Final Hurdle Before Global First CRISPR Gene-Editing Therapy Approval: FDA Advisory Committee to Review Exa-cel’s Off-Target Safety Data

CRISPR Therapeutics
Gene Editing Drug Developer

Vertex
Breakthrough Small Molecule Drug Developer
A Historic Moment in Gene Editing Is Approaching.
Recently, the FDA released a document announcing that it will convene an Advisory Committee (AC) meeting on October 31, requesting its external advisors on the Cellular, Tissue, and Gene Therapies Advisory Committee to discuss a key issue regarding exa-cel (exagamglogene autotemcel), a CRISPR-based gene-editing therapy developed by Vertex (Vertex Pharmaceuticals) and CRISPR Therapeutics:Evaluate whether the off-target analysis provides sufficient evidence to demonstrate its safety and determine if further studies are required.
According to the briefing documents released by the FDA,Although the FDA raised no concerns regarding the efficacy of exa-cel, stating that “even limitations in study design do not call into question the efficacy of exa-cel,” it questioned whether Vertex had used a sufficient sample size for off-target analysis.
On June 8 this year, Vertex and CRISPR Therapeutics jointly announced that the FDA had accepted the Biologics License Application (BLA) for exa-cel for the treatment of sickle cell disease (SCD) and transfusion-dependent beta-thalassemia (TDT). The PDUFA dates for the two indications are December 8, 2023 (SCD) and March 30, 2024 (TDT), respectively. If approved, exa-cel will become the world’s first approved CRISPR gene-editing therapy.
With the PDUFA date for exa-cel’s indicated use in sickle cell disease (SCD) approaching, this Advisory Committee (AC) meeting has effectively become the final hurdle for the drug’s market approval.And the final hurdle brings us back to the initial skepticism surrounding CRISPR gene-editing technology: Is CRISPR truly safe?
$1.1 Billion, Aiming for the World’s First CRISPR Gene-Editing Drug
In 2015, Vertex partnered with CRISPR Therapeutics to jointly develop gene therapies for hemoglobinopathies.
In 2017, Vertex and CRISPR Therapeutics announced the joint development of CTX001 (exa-cel).
In April 2021, Vertex announced updated terms of its collaboration with CRISPR Therapeutics regarding the investigational therapy CTX001 (exa-cel). Under the amended agreement, Vertex will lead the global development, manufacturing, and commercialization of exa-cel.CRISPR will receive a $900 million upfront payment, with potential milestone payments of $200 million upon the first regulatory approval of exa-cel.
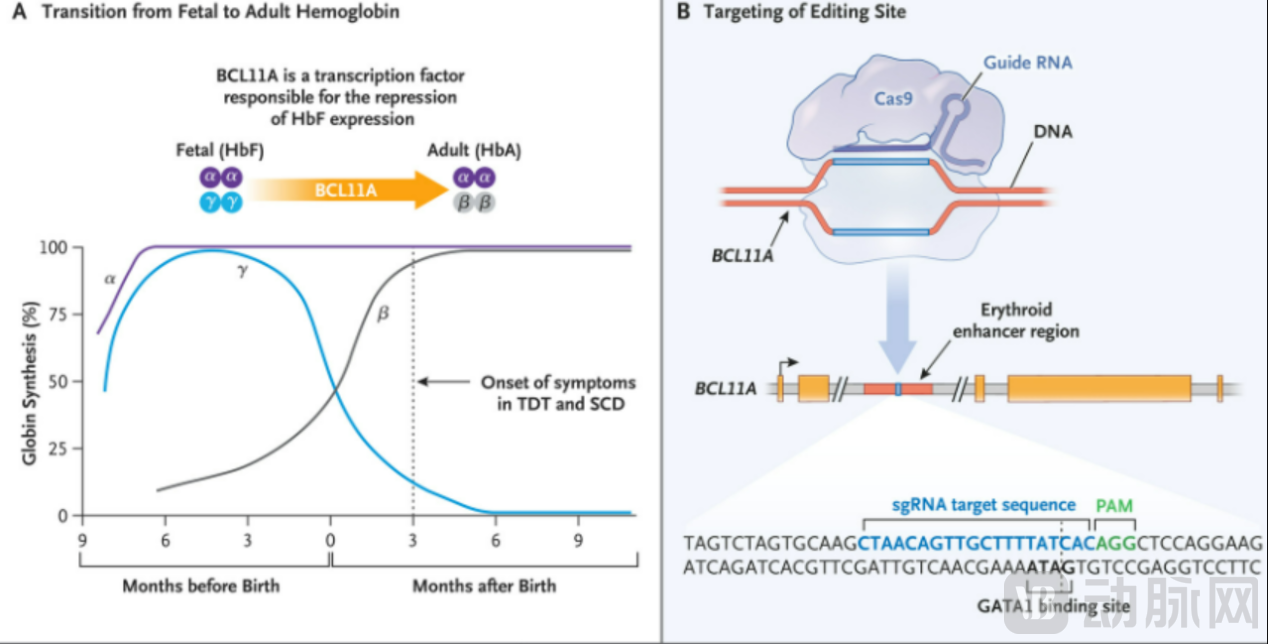
Vertex’s exa-cel, a CRISPR gene-editing drug that commanded an investment of $1.1 billion, works by using the CRISPR/Cas9 gene-editing system to edit patients’ own CD34+ hematopoietic stem cells ex vivo, specifically silencing the BCL11A enhancer (BCL11A is a transcription factor that inhibits the expression of γ-globin and fetal hemoglobin [HbF] in erythroid cells). This reactivates HbF production and leads to high-level HbF expression, which then transitions to adult-form hemoglobin, thereby treating sickle cell disease (SCD) and transfusion-dependent β-thalassemia (TDT).
In specific treatments, the patient’s own stem cells are collected, edited using CRISPR-Cas9 technology to target genetic defects, and finally infused back into the patient to treat the disease.This therapy requires only a single infusion to effectively treat both sickle cell disease (SCD) and transfusion-dependent β-thalassemia (TDT).
As the most advanced CRISPR gene-editing therapy currently in development, exa-cel has been granted Regenerative Medicine Advanced Therapy (RMAT) designation, Fast Track designation, Orphan Drug designation, and Rare Pediatric Disease designation by the U.S. FDA for the treatment of SCD and TDT. In Europe, the EMA and MHRA accepted the Marketing Authorization Application (MAA) for exa-cel in January 2023, also granting it Orphan Drug designation and Priority Medicines (PRIME) status. In the United Kingdom, exa-cel has additionally been awarded an Innovation Passport.
Currently, exa-cel is undergoing multiple clinical trials and long-term follow-up studies simultaneously. Focusing specifically on the sickle cell disease (SCD) indication that may receive approval in the near term, interim analysis disclosed in June showed that among 35 SCD patients treated with exa-cel, 17 were evaluable for primary and secondary endpoints, with no serious adverse events related to exa-cel observed. Among these 17 patients, approximately 94.1% achieved the primary endpoint (freedom from vaso-occlusive crises [VOC] for at least 12 consecutive months) (95% confidence interval: 71.3%, 99.9%; P=0.0001), with a mean VOC-free duration of 18.7 months and a maximum of 36.5 months; approximately 100% of patients achieved the secondary endpoint (freedom from VOC-related hospitalizations for at least 12 consecutive months) (95% confidence interval: 80.5%, 100.0%; P<0.0001).
In the analysis of all patients with sickle cell disease (SCD) treated with exa-cel, the mean fetal hemoglobin as a percentage of total hemoglobin exceeded 30% at Month 3, was maintained at approximately 40.0% during follow-up, and was associated with pancellular distribution.Over time, the edited BCL11A tends to stabilize in the bone marrow and peripheral blood.
exa-cel vs. lovo-cel: Which Will Capture the Market First?
Sickle Cell Disease (SCD) is a blood disorder caused by an inherited abnormal hemoglobin (the oxygen-carrying protein in red blood cells), leading to severe pain and organ damage, including the kidneys, cardiopulmonary system, and brain.
Currently, the mainstream treatments for early-stage sickle cell disease (SCD) include hydroxyurea (a chemotherapy agent approved in 1998) and red blood cell transfusions. Approved drugs such as L-glutamine, Voxelotor, and Crizanlizumab are also available to improve the prognosis of SCD patients. Allogeneic hematopoietic stem cell transplantation (HSCT) remains the only curative therapy currently available for SCD; however, according to data disclosed by the FDA, this approach carries significant risks, and fewer than 20% of SCD patients can find a suitable human leukocyte antigen (HLA)-matched donor. Consequently, there remains a substantial unmet clinical need in the management of SCD.
The brave prevail when paths cross. In the field of sickle cell disease (SCD), bluebird bio’s gene therapy lovo-cel (lovotibeglogene autotemcel) also demands attention. Lovo-cel is a one-time gene therapy designed to add a copy of a modified β-globin gene to the patient’s extracted hematopoietic stem cells.In April this year, Bluebird Bio announced that it had submitted a Biologics License Application (BLA) for the gene therapy lovo-cel to the FDA, with a PDUFA date of December 20.
In comparison, both exa-cel and lovo-cel achieved maximum hemoglobin increases exceeding 4 g/dL. With follow-up approaching two years to date, 100% of patients have restored their hemoglobin levels to normal, indicating freedom from transfusion dependence. Meanwhile, both therapies demonstrated excellent efficacy in preventing vaso-occlusive crises (VOCs); patients remained free from pain and VOC crises for one year, with no VOC-related hospitalizations reported among these patients.
However, the two therapies differ in their safety profiles. The clinical development of lovo-cel, a lentiviral vector-based gene editing therapy, was once suspended for an extended period after two patients developed acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), which are malignant hematologic disorders. Additionally, 17% of patients experienced delayed platelet engraftment, and platelet levels in all patients failed to return to pre-treatment baselines. For exa-cel, the longest follow-up in patients with sickle cell disease (SCD) has reached 36.5 months, during which they have remained free of vaso-occlusive crises (VOCs); however, data on hemoglobin maintenance in the earliest enrolled patients have not been disclosed.
Furthermore, the CRISPR gene-editing programs of companies such as Beam Therapeutics and Editas Medicine are all in Phase I clinical trials.
Off-Target Effects: One of the Key Issues Regarding CRISPR Safety
According to documents previously disclosed by the FDA, off-target effects of CRISPR are a key issue currently under review for exa-cel. Since the emergence of CRISPR gene-editing technology in 2012, off-target effects have been one of the major constraints on its development. Incomplete pairing between gRNA and target DNA sequences can lead to unintended editing across the genome, causing mutations. Such off-target mutations may disrupt the function or regulation of non-target genes, leading to serious consequences.
Therefore, during the optimization of CRISPR technology, editing of cells, and development of therapies, it is essential to optimize various strategies—such as careful selection of target sites, optimization of sgRNA design and Cas9 activity, and off-target detection analysis—to minimize the safety risks associated with off-target effects. Furthermore, to definitively assess the efficacy of CRISPR and its long-term in vivo consequences, quantitative bioinformatics methods leveraging whole-genome next-generation sequencing (NGS) data are required to screen for unintended genomic edits.
According to the FDA’s preliminary disclosure, Vertex utilized variant information from the 1000 Genomes Project database, which contains sequencing data from 2,504 individuals. By analyzing variants with an allele frequency greater than 1% in at least one continental population group within this database, 50 additional off-target candidate sites were identified.
Meanwhile, the reference database used by Vertex contains 661 whole-genome sequencing (WGS) datasets from the target population. This set includes 61 sequencing datasets from the southwestern United States. The FDA has stated that it remains unclear whether this limited number of WGS datasets is sufficient to capture the variants present in the target population, and insufficient sequencing data may hinder the identification of relevant variants associated with off-target editing.
For off-target analysis in cells, Vertex used three samples from healthy donors and three samples from subjects with sickle cell disease (SCD) of African ancestry. The FDA stated that “given the impact of SCD on hematopoietic stem and progenitor cell (HSPC) function, which may alter the chromatin landscape and potentially affect off-target editing, the advantages of using healthy donor samples for such analyses are unclear. Furthermore, it remains uncertain whether the small number of samples used in the cellular guide sequence off-target analysis is sufficient to adequately assess off-target editing ex vivo.”
Luca Issi, an analyst at RBC Capital Markets, described the documents disclosed by the FDA as “benign,” writing in a client note that the agency’s focus appeared to be on “technical [...] issues rather than broader risks for patients receiving this therapy.” Salim Syed of Mizuho Securities shared a similar view, suggesting the documents could represent a “best-case scenario.” He also noted that the FDA had merely posed a “discussion question” to the advisory committee experts, rather than calling for a recorded vote.
According to the FDA announcement, the meeting will be held on Tuesday (Eastern Time), with participation from 14 experts, consumers, and patient representatives in the fields of gene editing, stem cell biology, biostatistics, and sickle cell disease. It is worth noting that while the FDA follows the committee’s recommendations, they are not binding.