Will the $3.2 Million Gene Therapy Elevidys Be Withdrawn? Sarepta Submits New Data Amid FDA Scrutiny

Sarepta
Developer of Therapies for Rare Neuromuscular Diseases
On October 30, Sarepta Therapeutics announced preliminary data from EMBARK (Study SRP-9001-301), the Phase 3 confirmatory trial of Elevidys, the world’s first one-time gene therapy for Duchenne Muscular Dystrophy (DMD).The study did not reach the primary clinical endpoint of NSAA score (North Star Ambulatory Assessment, a measure of motor function).
In June this year, Elevidys received FDA approval for marketing, indicated for ambulatory children aged 4–5 years with Duchenne muscular dystrophy (DMD) (contraindicated in DMD patients with deletion mutations in exon 8 and/or exon 9). Subsequently, this new drug, priced at $3.2 million per dose (approximately RMB 23.41 million), quickly ranked among the most expensive medications, becoming the second-most costly drug worldwide.
However, based on the data available at the time, Sarepta’s initial goal was to target Elevidys for patients of all age groups, whereas the FDA’s accelerated approval restricted its use to children aged 4–5 years. This clinical trial aimed, first, to confirm the clinical benefit of Elevidys, and second, to engage in discussions with the FDA to expand the eligible population. According to Elevidys’ subsequent response in a public letter, the company hopes to initially expand the age range to 4–7 years, and further broaden indications in the future with respect to disease stage, antibody status, and types of gene mutations.
Duchenne muscular dystrophy (DMD) is a common X-linked recessive muscular dystrophy, with an incidence of approximately 1 in 3,500 male newborns. Genetic testing has identified a genetic etiology in 93.1% of patients, laying the foundation for early gene therapy. Due to genetic mutations, patients lack dystrophin in their muscle tissue, leading to muscle weakness and progressive muscle loss. Most affected children lose the ability to walk by age 12 and typically die around the age of 30.
Prior to the approval of Elevidys, clinical management of Duchenne muscular dystrophy (DMD) primarily relied on symptomatic and supportive treatments, such as corticosteroids and hormone replacement therapy. However, these therapies require long-term administration and can only alleviate disease symptoms without addressing the underlying cause.
Elevidys is a gene replacement therapy co-developed by Roche and Sarepta. It addresses the root cause of Duchenne muscular dystrophy (DMD) by targeting the production of functional components of dystrophin in muscle tissue. The therapy involves packaging a transgene encoding a truncated form of dystrophin (micro-dystrophin) into an adeno-associated virus (AAV) vector. A single intravenous infusion enables patients’ muscles to produce a recombinant protein with partial dystrophin function, making it effective for patients harboring any type of DMD-causing genetic variant.
In May this year, the FDA advisory committee voted 8-6 in favor of the accelerated approval of Elevidys. In June, the FDA approved Elevidys as the world’s first gene therapy for Duchenne muscular dystrophy (DMD). However, because Sarepta used micro-dystrophin expression levels as a surrogate endpoint, the FDA required the completion of confirmatory clinical trials to verify clinical benefit.
This confirmatory clinical trial is a global, randomized, double-blind, placebo-controlled Phase 3 study targeting DMD patients aged 4 to 7 years. According to Sarepta’s disclosure, the trial enrolled a total of 125 patients, who were randomized in a 1:1 ratio to receive either Elevidys or placebo. The primary endpoint was the North Star Ambulatory Assessment (NSAA) score, while secondary endpoints included protein expression levels, time to rise (TTR), and the 10-meter walk/run test (10MWR).
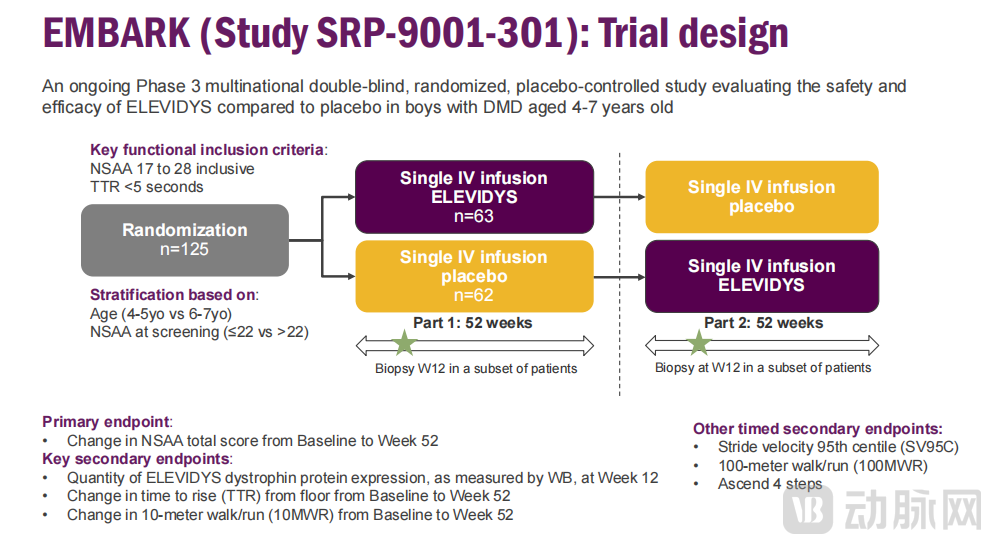
Data show that in the EMBARK trial, participants treated with Elevidys demonstrated an improvement in NSAA scores at Week 52 compared to those receiving placebo; however, the prespecified primary clinical endpoint was not met (improvement showed a trend but did not reach statistical significance).
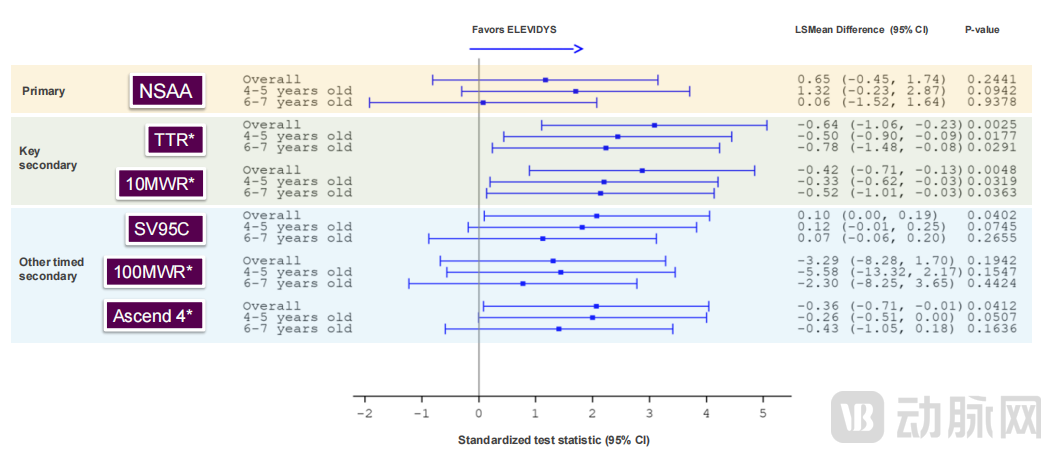
Meanwhile, all key prespecified secondary endpoints, including time to rise (p=0.0025) (TTR) and the 10-meter walk/run test (p=0.0048) (10MWR), demonstrated robust and statistically significant improvements, providing evidence of clinically meaningful therapeutic benefits, with statistical significance observed across all age groups.
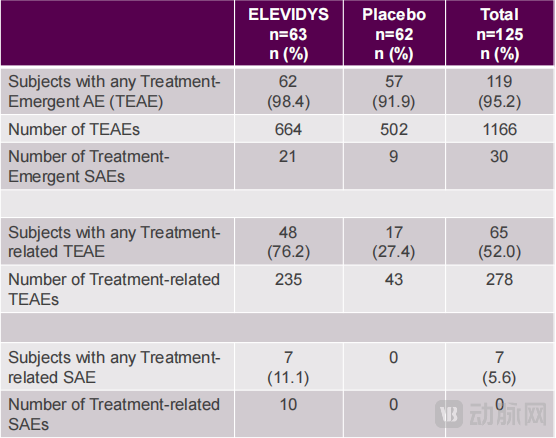
Furthermore, no new safety signals were observed in the EMBARK trial. Among 125 subjects, only 7 experienced treatment-related serious adverse events, with severity consistent with previous experimental data.
In 2019, Roche acquired the exclusive distribution rights for Elevidys outside the United States for $1.15 billion. Under the agreement, Roche paid Sarepta a $750 million upfront payment and invested $400 million to subscribe to its shares. Upon approval of Elevidys and achievement of certain sales milestones, Sarepta could receive up to $1.7 billion in additional payments.
This rare-disease drug, in which Roche has placed high hopes, had previously received FDA Fast Track designation and Rare Pediatric Disease (RPD) designation, as well as Orphan Drug Designation (ODD) in the United States, the European Union, Switzerland, and Japan.
The accelerated approval of Elevidys was based on the increased expression of micro-dystrophin in skeletal muscle. According to public information, Sarepta was the first to file for approval after its Phase 2 clinical trial met the clinical endpoints. The submitted Biologics License Application (BLA) included efficacy and safety data from three clinical studies, as well as a comprehensive analysis of these three studies, comparing functional outcomes with propensity score-matched external controls (EC).
Generally, if a clinical trial fails to meet its primary endpoint, the FDA will not consider data from secondary study objectives as positive. In other words, under the FDA’s Accelerated Approval pathway, a drug may be approved for marketing based on single-arm clinical trials and surrogate endpoints to address urgent unmet clinical needs, provided that two conditions are met: first, the sponsor must complete the agreed-upon confirmatory clinical trials within the timeframe specified in the agreement with the FDA; second, if the anticipated benefit-risk profile is confirmed, the FDA will convert the accelerated approval into full approval; otherwise, the accelerated approval will be withdrawn.
According to a 2021 study published in The BMJ, nearly half of the 253 drugs approved since the FDA established its Accelerated Approval pathway in 1992 have not been confirmed as clinically effective. Further analysis of FDA data revealed that only 16 drugs approved through this pathway have been withdrawn, with most withdrawals due to demonstrated lack of efficacy; in some cases, confirmatory trials had not even been conducted.
In recent years, the FDA’s accelerated approval pathway has been contracting due to the failure of several innovator drugs granted accelerated approval in their confirmatory Phase III clinical trials. With Robert Califf returning for a second term as FDA Commissioner, a new framework—including provisions for accelerated withdrawal—is on the horizon.
Nevertheless, Sarepta has expressed a positive outlook on the results of the EMBARK trial. According to Stat, Sarepta has met with senior FDA officials, including the Director of the Center for Biologics Evaluation and Research (CBER). Doug Ingram, CEO of Sarepta, stated, “The FDA will rapidly review the new data and complete its assessment of the label expansion for the indication.”
In light of this, claims that the drug would be “withdrawn” are likely baseless. Sarepta subsequently addressed questions regarding the future expansion of Elevidys’ indications in an open letter to its community, covering aspects such as age, disease stage, antibody status, and types of genetic mutations. Key points include:
·In addition to the currently FDA-approved indications and the population covered by the EMBARK study, Sarepta is conducting a global Phase 3 clinical trial, ENVISION (NCT05881408), aimed at evaluating the safety and efficacy of Elevidys in ambulatory boys (of any age) and non-ambulatory older boys (aged 8–18 years).
·Two distinct studies are expected to be initiated to explore two different mechanisms for reducing pre-existing AAVrh74 antibodies.