CRISPR/Cas9 Therapy Casgevy (exa-cel) Gains FDA Approval: A Milestone for Gene Editing and Commercialization

CRISPR Therapeutics
Gene Editing Drug Developer
On December 8 (local time), Vertex Pharmaceuticals and CRISPR Therapeutics jointly announced that the U.S. Food and Drug Administration (FDA) has approved Casgevy (exa-cel), a CRISPR/Cas9 genome-edited cell therapy co-developed by the two companies, for marketing. It is indicated for the treatment of patients aged 12 years and older with sickle cell disease (SCD) who experience recurrent vaso-occlusive crises (VOC).
Last month, exa-cel received conditional marketing authorization from the UK Medicines and Healthcare products Regulatory Agency,It became the world’s first approved CRISPR gene-editing therapy. As the globally recognized standard-setter, FDA approval often signifies that a product has more reliable endorsement and broader market potential, marking a milestone starting point for the true commercialization of CRISPR therapies.
The FDA has been striving to strike a balance between efficacy and safety for cutting-edge therapies such as cell and gene therapies (CGT). Recently, in response to multiple adverse event reports associated with CAR-T therapies, the FDA announced that it is investigating cases of malignancies observed in patients receiving marketed CAR-T therapies in the United States and assessing the need for regulatory actions, thereby reigniting debate in this field. This serves as a reminder to pharmaceutical companies and biotech firms that regulatory scrutiny on safety will not be relaxed, even when clinical benefits are significant.
The same applies to CRISPR therapies; until late October, the FDA was still convening an Advisory Committee (AC) meeting to assess the risk of off-target effects associated with exa-cel gene editing.
This FDA approval undoubtedly marks a significant milestone in the history of human technology. It represents a new achievement for Vertex, leveraging its robust operational capabilities to advance in the fields of rare diseases and gene therapy, thereby paving the way for the further development of CRISPR and gene therapies.
How Did Vertex and CRISPR Therapeutics “Catch Up and Take the Lead”?
Exa-cel is not the only therapy targeting sickle cell diseaseGene Therapy.On the same day, Lyfgenia (lovo-cel) from veteran gene therapy company Bluebird Bio also received FDA approval for the treatment of patients aged 12 years and older with sickle cell disease (SCD) and a history of vaso-occlusive events (VOE).
Bluebird’s product works by using a lentiviral vector to deliver a functional copy of the β-globin gene into patients’ hematopoietic stem cells, thereby enabling the production of normal hemoglobin.
Bluebird began in 2016lovo-celdevelopment entered Phase 1/2 clinical trials and received Breakthrough Therapy Designation from the FDA in December 2018. In July 2019, bluebird bio announced the initiation of a Phase 2/3 clinical trial, named the HGB-210 study, to continue evaluating the safety and efficacy of lovo-cel.
In comparison, Vertex and CRISPR Therapeutics were latecomers. Around 2013, Bluebird Bio’s core technology—ex vivo editing using lentiviral vectors—was highly sought after, while CRISPR Therapeutics had just been established. In 2016, CRISPR Therapeutics announced a collaboration with Vertex to develop exa-cel (then named CTX001). However, in May 2018, the FDA halted the clinical trial of exa-cel for sickle cell disease before it had even begun.
However, as one of the most successful biotech companies in history, Vertex is well-versed in addressing the various challenges associated with new drug development.Vertex spent 18 years developing the hepatitis C drug telaprevir and debated its side effects with the advisory committee at an FDA hearing under intense pressure, ultimately securing unanimous approval.
After multiple rounds of discussion and the submission of additional data, the FDA lifted the clinical hold on exa-cel five months later.
CRISPR Therapeutics has had strong partners from the outset. Subsequently, exa-cel progressed smoothly through development and entered Phase 3 clinical trials in 2022. At the European Hematology Association Congress held in June of that year, Vertex and CRISPR Therapeutics announced that seven patients with sickle cell disease remained free from transfusions for up to 21 months following treatment with exa-cel.
Over the past few years, exa-cel has secured nearly all major regulatory designations for innovative drug development. In the United States, the FDA granted exa-cel Regenerative Medicine Advanced Therapy (RMAT), Fast Track, Orphan Drug, and Rare Pediatric Disease designations for the treatment of sickle cell disease (SCD) and transfusion-dependent beta-thalassemia (TDT). In Europe, the European Medicines Agency (EMA) and the UK’s Medicines and Healthcare products Regulatory Agency (MHRA) accepted the Marketing Authorization Application (MAA) for exa-cel in January 2023, also granting it Orphan Drug designation and Priority Medicines (PRIME) status. In the UK, exa-cel was additionally awarded the Innovative Licensing and Access Pathway (ILAP) “Innovation Passport.”
But Bluebird’s lovo-cel was not so fortunate.In November 2020, the FDA delayed the marketing application for lovo-cel by a full year due to CMC-related issues. In February of the following year, the FDA imposed its first clinical hold on lovo-cel trials after two enrolled patients developed suspected hematologic malignancies. Just five months after the hold was lifted, the FDA partially suspended lovo-cel clinical studies again in late 2021 due to anemia observed in treated patients.

Reasons for the FDA Placing Clinical Studies on Clinical Hold
This has significantly impacted Bluebird’s progress. As planned, Bluebird had aimed to submit a Biologics License Application (BLA) to the FDA by the end of the first quarter of 2023, but ultimately failed to do so.Instead, Vertex and CRISPR Therapeutics were the first to complete their rolling Biologics License Application (BLA) submissions on April 4, 2023, while bluebird bio did not submit its BLA until April 24, thereby losing its “first-place” distinction.
Setting aside operational capabilities, the competition between Exa-cel and lovo-cel is, first and foremost, a contest between two distinct technological approaches.A major drawback of lentiviral vectors is the risk of insertional mutagenesis leading to oncogene activation, a challenge that remains difficult to resolve. In contrast, the primary concern surrounding CRISPR technology is off-target effects; however, these risks can be minimized through various optimization strategies, such as careful selection of target sites, optimization of sgRNA design and Cas9 activity, and off-target detection analyses.
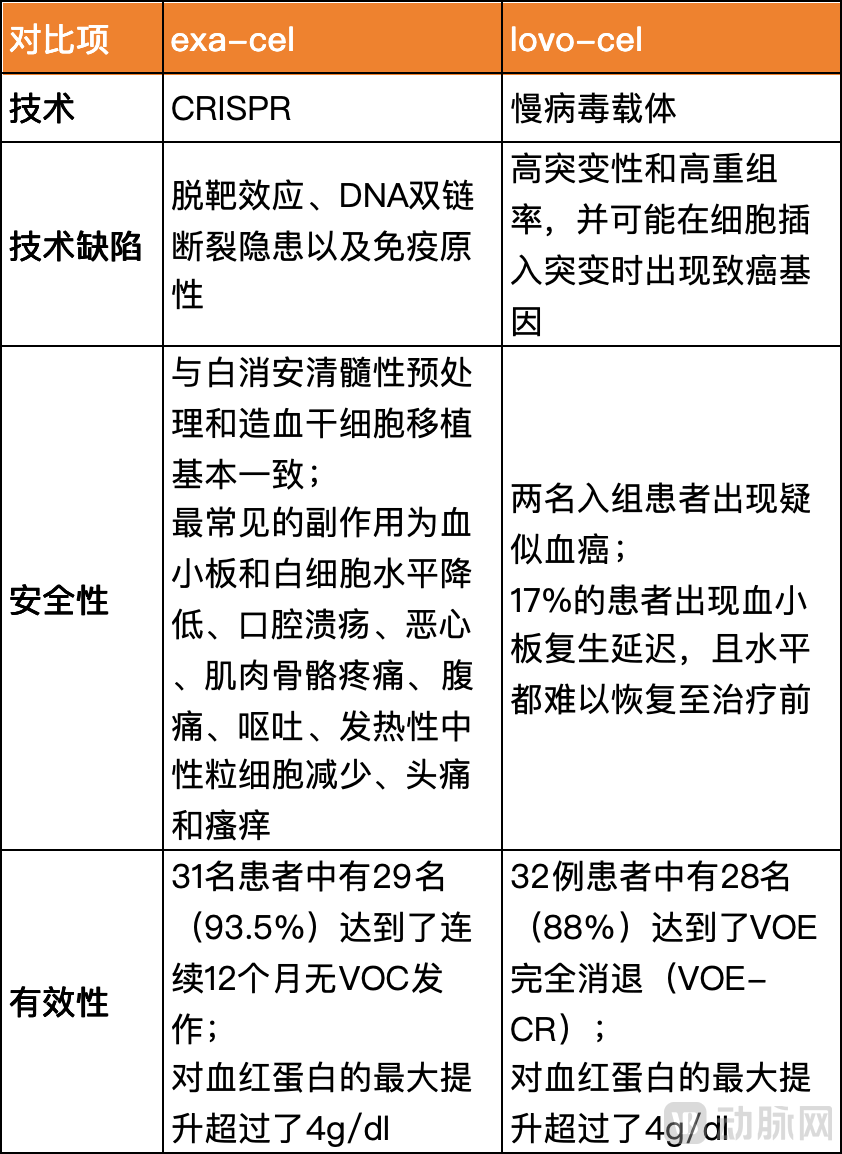
In terms of safety, this demonstrates how next-generation technologies are decisively outperforming existing ones. The development of Bluebird Bio’s gene therapies has been quite tortuous; although there are already marketed products, lentiviral technology continues to face skepticism, while gene-editing technologies such as CRISPR are advancing rapidly, gradually capturing the majority of market attention and funding.
Can Vertex Make Money from Gene Therapy?
As the protagonist of *The Billion-Dollar Molecule*, Vertex is renowned for its aggressive style, high-caliber team, and good fortune. With a market capitalization now approaching $100 billion, Vertex can be regarded as a leading innovator in innovative drugs, resilient to economic cycles. According to this year’s financial reports, the third-quarter performance guidance has been raised to nearly $10 billion, excluding the anticipated sales from exa-cel.
Historically, Vertex’s star hepatitis C drug Incivek and its cystic fibrosis drugs Symdeko and Kalydeco have generated substantial returns for the company, even though none of these therapies originated from Vertex’s internal pipeline.Vertex has two major strengths: an elite business development (BD) team and the boldness to truly translate science into commercial success.
In the collaboration between Vertex and CRISPR Therapeutics, Vertex takes the leadexa-celthe global development, manufacturing, and commercialization; initially, the two parties shared R&D costs and profits equally, which was later revised to a 60/40 split in favor of Vertex and CRISPR Therapeutics, respectively.
Commercialization has always been a major challenge for gene therapies. Bluebird Bio already has two marketed gene therapies, but their commercialization has not gone smoothly due to high prices. To alleviate payers' concerns, Bluebird Bio has adopted an outcomes-based payment model, namely "full upfront payment with refund if treatment fails," yet this has still failed to improve the company's cash flow situation.
In contrast, the potential commercial advantages of exa-cel include:
- CRISPR technology offers generational advantages in CMC and cost. Vertex directly reduced the price of exa-cel to $2.2 million, below Bluebird’s lovo-cel at $3.1 million.
- Although SCD is a rare disease, it affects a relatively large patient population. Similar to Novartis’s Zolgensma, a profitability benchmark for gene therapies, whose indication—spinal muscular atrophy—is one of the more common rare diseases with a substantial number of patients, exa-cel’s demonstrated efficacy and safety profile will likely attract a certain number of patients willing to pay for the treatment.
- Vertex is a company sustained by rare disease drugs. It clearly possesses extensive experience in monetizing therapies within the rare disease space and knows how to drive non-in-house products to success. Having navigated complete drug life cycles and multiple capital market cycles, it boasts competitive capabilities that are difficult for typical biopharmaceutical companies to match.
- From Vertex’s investment perspective, it has placed significant bets on CRISPR and the broader cell and gene therapy (CGT) sector. Through acquisitions and joint development initiatives, Vertex has established a multi-faceted presence in gene editing technologies, delivery mechanisms, and therapeutic indications. This strategy echoes the words of its founder, James Barksdale, who once wrote, “We don’t fear hard work or failure; we truly fear mediocrity.” With such foresight and determination, Vertex is poised to position CRISPR and more advanced gene therapies as its future growth engines.
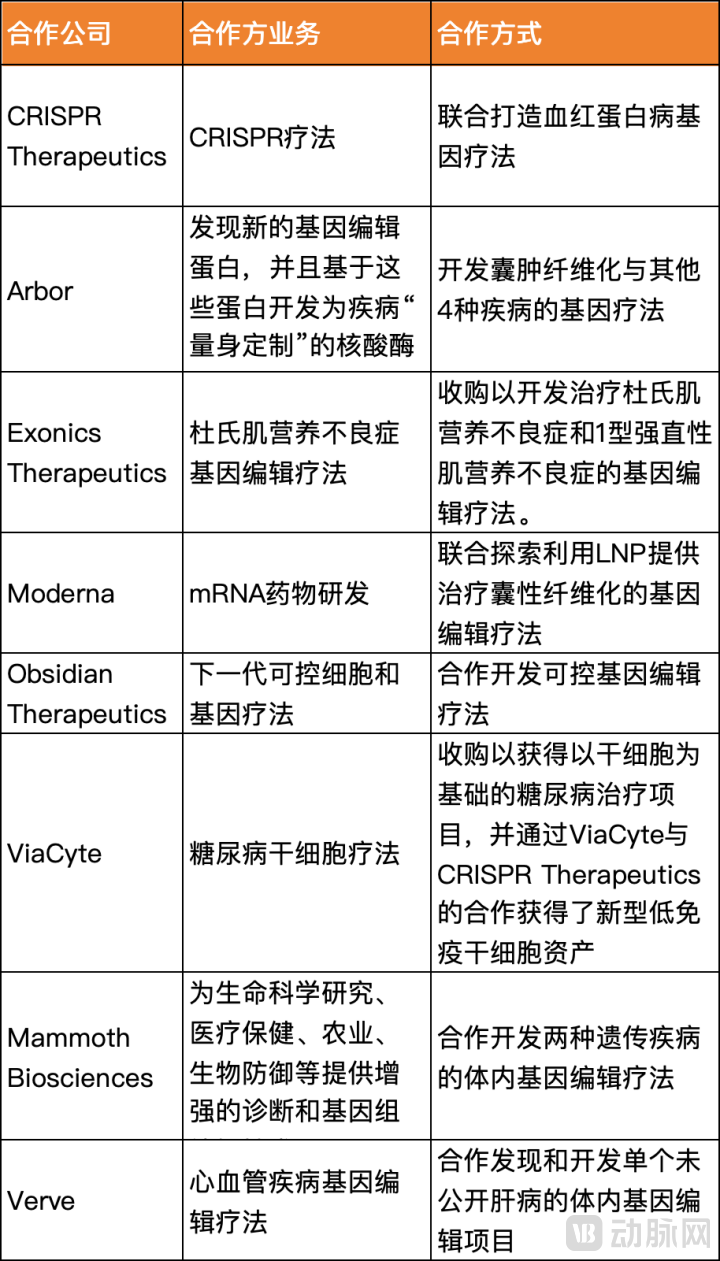
Vertex’s External Collaborations in Gene Therapy
The Road Ahead for CRISPR: Breadth and Accessibility
Regarding the FDA’s approval of exa-cel, some investors stated: “The CRISPR therapy approved this time remains an ex vivo editing approach. More accurately, this personalized treatment should be regarded as a specific application of gene editing as a tool, whereas the next generation of in vivo gene editing will more closely resemble a universal ‘drug.’”
In vivo gene editing covers more indications, target cells, and target organs; it has a better cycle, with products in universal drug forms such as nanoparticles or viral vectors, eliminating the need for personalized cell preparation processes; most importantly, the treatment process is more controllable. Vertex’s investment portfolio also includes assets in in vivo gene editing, and among the many CRISPR therapies currently in active development worldwide, there are numerous in vivo editing projects.
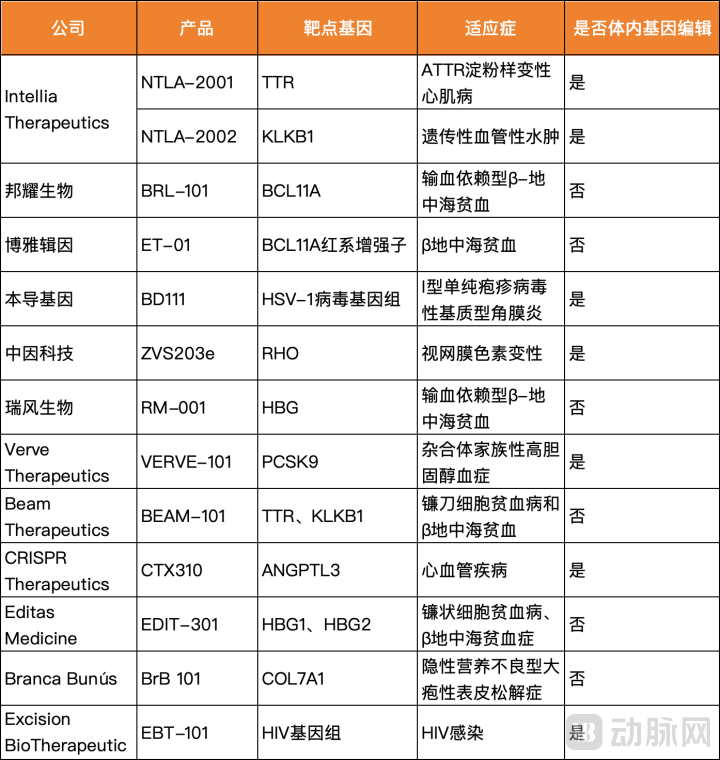
Some industry insiders believe that in vivo gene editing will be the trend, but the FDA currently maintains a highly cautious stance toward this technology. Pioneering companies in in vivo editing, such as Intellia and Verve, have had their programs halted by the FDA due to safety concerns. For instance, Verve-101, a base-editing therapeutic, was only approved by the FDA last month to enter clinical trials in the United States after a one-year clinical hold.
However, in the gene editing report released by Evaluate Vantage this year, multiple globally leading gene therapy companies stated that both ex vivo and in vivo editing will have their respective room for development.
“There are still many issues to be resolved in the translation of in vivo gene editing from animal models to human applications. The advantages and disadvantages of in vivo and ex vivo gene editing should not be compared in isolation, nor should it be simply assumed that the cost of in vivo gene editing is necessarily lower than that of ex vivo editing. Instead, evaluation and selection should be conducted from multiple perspectives based on the specific disease and application scenario.” In this regard,Dr. Zheng Biao, CEO of Bonito Biostated to VCBeat.
Chinese cell and gene therapy (CGT) companies, represented by Bangyao Biotech and Ruifeng Biotech, have established another global hub for gene therapy, a trend that warrants close attention. Notably, Chinese firms account for nearly half of the CRISPR-based therapies currently in development.
For example, BRL-101, the CRISPR gene-editing therapeutic product developed by Biocytogen for transfusion-dependent β-thalassemia, is poised to enter Phase II clinical trials shortly. The first pediatric patient with severe thalassemia treated with BRL-101 gene therapy has remained free from transfusion dependence for over three years, marking the world’s first successful case of treating β0/β0 severe thalassemia using CRISPR gene-editing technology. This achievement represents a breakthrough in clinical research on gene-editing therapies in China.
RayWind Biosciences Recently Announced Clinical Phase I Progress of RM-001, a Gene-Editing Therapy for Transfusion-Dependent β-Thalassemia: All 7 Patients in the Investigator-Initiated Trial (IIT) and 9 Patients Enrolled in the Investigational New Drug (IND) Phase I Trial Successfully Achieved Transfusion Independence Following Treatment, Paving the Way for the Upcoming Phase II Clinical Trial.
BenDaoGene’s BD111 injection received U.S. FDA approval for its clinical trial application in July, with an indication for herpes simplex virus type 1 (HSV-1) stromal keratitis, marking it as the world’s first CRISPR-based antiviral orphan drug approved by the FDA.
Dr. Liang Junbin, CEO of Ruifeng Bio“Speaking to VCBeat, [the source] stated: ‘The approval of CRISPR therapies will further drive research and development in gene therapy within China, particularly by promoting the support and refinement of the regulatory framework for this class of drugs. The commercialization of gene-editing medicines will prompt relevant authorities to establish more comprehensive guidance documents to ensure safety and efficacy, which will also help standardize the research of other gene therapies in China.’”
Another advantage of Chinese companies is their ability to innovate by leveraging more cost-effective clinical and CMC solutions, thereby enhancing the accessibility of gene therapies.
Dr. Zheng BiaoIntroduction: “In China, we can strive to control costs across all stages, including raw materials, labor, and CMC processes. We are actively working to localize key consumables. For instance, Bangyao Biotech has entered into a strategic partnership with Porton Biopharma to leverage its manufacturing advantages for commercial-scale production. We also clearly recognize the support provided by China’s national medical insurance policies for the rare disease sector, and we believe that innovative therapies will benefit more patients in the future.”
Dr. Liang JunbinHe also remarked, “The cost of gene therapy is determined not only by manufacturing expenses but also by multiple factors, including R&D expenditures, clinical trial costs, regulatory fees, and patent costs. Overall, China’s cost advantage is anticipated; however, improving the accessibility of gene therapies also depends on payment systems such as national medical insurance and commercial insurance, supportive policy frameworks, consensus and guideline development for indications, drug distribution capabilities, and GMP manufacturing capacity. Crucially, it is essential to provide innovative pharmaceutical products, such as gene therapies, with commensurate value recognition and commercial space, thereby fostering a virtuous cycle that benefits more patients and cures more diseases.”