China Launches 'Green Channel' for Importing New Drugs, Allowing Priority Domestic Approval Post-Clinical Trial
Despite ongoing reforms to the drug review and approval system, the “major move” recently unveiled by the China Food and Drug Administration (CFDA) has still caught the industry somewhat off guard.
Recently, the China Food and Drug Administration (CFDA) issued a decision on the registration management of imported drugs, proposing to allow drugs that are unregistered or have not yet entered clinical trials abroad to conduct international multi-center clinical trials in China. After completing clinical trials in China, applicants may directly submit applications for drug marketing registration.
This effectively opens a “green channel” for imported drugs, significantly narrowing the gap between their launch timelines abroad and in China. The era when multinational pharmaceutical companies had to wait years for their new drugs to enter the Chinese market will become a thing of the past. By breaking down the “policy barriers” in the approval and review process, domestic pharmaceutical companies will truly compete on a level playing field with multinational corporations, carrying profound implications.

The New Policy Makes Significant Cuts
According to the document titled “Decision on Adjusting Certain Matters Concerning the Registration and Administration of Imported Drugs (Draft for Comments)” (hereinafter referred to as the “Draft”), obtained by VCBeat (WeChat ID: vcbeat), the primary objective of this round of policy adjustments is to “encourage the simultaneous conduct of clinical trials domestically and abroad for new drugs not yet marketed overseas, upon approval, thereby shortening the time lag between their market launches in China and other countries, and meeting the public’s clinical needs for new medicines.”
According to the “Opinions,” clinical trials and marketing approval of imported drugs will be adjusted as follows:
First, for international multi-center drug clinical trials conducted in China, the requirement that investigational drugs must have been registered overseas or have already entered Phase II or Phase III clinical trials is abolished, except for vaccine products.
Second, for international multi-center drug clinical trials conducted in China, an application for drug marketing registration may be submitted directly upon completion of the trials. When submitting the marketing registration application, the requirements of the Measures for the Administration of Drug Registration and related documents shall be complied with.
Third, the requirement to obtain marketing authorization from the country or region where the overseas pharmaceutical manufacturer is located is waived for new chemical drugs and innovative therapeutic biological products applying for import.
Fourth, for registration applications submitted prior to the issuance of this Decision that rely on international multi-center clinical trial data to request a waiver of import clinical trials, importation may be approved if the requirements are met.
As can be seen from the above, the “Opinions” make substantial changes to the original regulatory measures for imported drugs.
According to VCBeat’s research, the previous Measures for the Administration of Imported Drugs stipulated that imported drugs undergoing clinical trials must have obtained registration approval from the drug regulatory authority in their country of origin or have already entered Phase II or later clinical trials; furthermore, drugs applying for market launch in China must have already obtained marketing authorization abroad.
The “Opinions” eliminate the requirements for clinical trials and overseas marketing authorization. In the future, foreign pharmaceutical companies can conduct international multi-center drug clinical trials in China simultaneously, and upon successful completion of these trials, they may even obtain market approval in China prior to overseas launch.
“This means that foreign pharmaceutical companies can develop new drugs specifically for the Chinese market, and China’s vast pharmaceutical market will open doors for these international firms,” Lu Sheng, a partner at Shengrui Consulting, told VCBeat.
Currently, multinational pharmaceutical companies account for approximately 20% of China’s pharmaceutical market, with constraints including prolonged review timelines and clinical trial processes.
“According to our survey data, the approval time for clinical trials of imported drugs generally exceeds six months, and the review period for production registration takes more than two years, resulting in a total timeline of up to three to five years. This has led to discrepancies in the market launch timelines of new drugs between China and other countries, with some patients unable to wait for access to certain new therapies and even opting to seek medical treatment abroad,” said Lu Sheng. He noted that the prolonged timeline for introducing imported drugs indeed partially hinders patients’ access to necessary medications.
Significant Progress in Drug Review and Approval System Reform
From the perspective of medication safety, drugs that have undergone multiple phases of clinical trials and obtained marketing approval abroad exhibit a higher safety profile. However, as China’s capabilities in conducting clinical trials and approving pharmaceuticals improve, transferring or front-loading the clinical trials and marketing approval processes for imported new drugs will help enhance the efficiency of drug review and reduce the waiting time for market entry by enterprises.
In fact, there have been significant changes in the review of new drugs in China over the past five years, gradually shifting from an administrative procedural approach to one oriented toward scientific justification. The supporting clinical trial capabilities and regulatory workforce have also developed accordingly.
“To ensure accuracy in clinical trials and prevent data fabrication, the China Food and Drug Administration (CFDA) launched a data-cleaning initiative targeting clinical trial centers. It also encouraged capable research institutions, universities, private organizations, and pharmaceutical companies to collaborate on conducting clinical trials. Coupled with the recently implemented Marketing Authorization Holder (MAH) system and the consistency evaluation for generic drugs—which both impose requirements on clinical centers—the number and scope of domestic clinical trial centers have increased by at least 30% compared to the period around 2012.” Lu Sheng believes that this enhanced clinical trial capacity is the primary reason why the CFDA has been emboldened to expand clinical trials for imported new drugs.
Previously, Bi Jingquan, then Commissioner of the China Food and Drug Administration (CFDA), acknowledged that a shortage of domestic regulatory reviewers had objectively constrained the speed at which imported drugs could enter the market. “Our drug review process has been relatively lengthy, and our review capacity has been limited. The U.S. Center for Drug Evaluation and Research (CDER) employs 5,000 staff, whereas we increased our workforce to 600 by the end of last year through concerted efforts. While efficiency has improved compared with earlier periods, a gap still remains. We are gradually enhancing approval efficiency by optimizing processes and strengthening our workforce. As a result, the backlog in drug reviews has significantly decreased from its peak of 22,000 applications to 8,000 by the end of last year.”
According to the “2016 Annual Drug Review Report” (hereinafter referred to as the “Report”), released on the same day by the China Food and Drug Administration (CFDA) alongside the “Opinions,” the Center for Drug Evaluation (CDE) improved review efficiency and quality through measures such as strengthening review project management, refining review procedures, enhancing timeline management, establishing special task forces, increasing the number of reviewers, and implementing tiered authorization for sign-off.
A notable change is that the backlog of drug reviews has been significantly cleared.
According to the Report, the Center for Drug Evaluation (CDE) completed the review and submitted 12,068 registration applications to the China Food and Drug Administration (CFDA) throughout last year. Review of 943 registration applications was completed but pending applicants’ responses to address deficiencies in the submission materials. The total number of registration applications reviewed during the year increased by 26% compared with 2015. The backlog of pending registration applications decreased from a peak of nearly 22,000 in September 2015 to 8,200, basically eliminating the registration backlog.
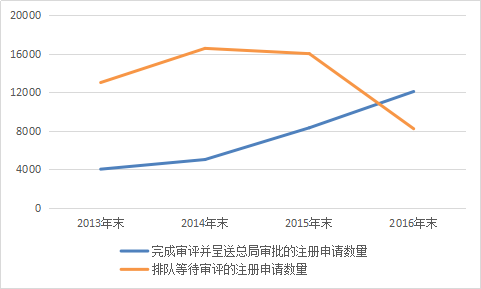
Comparison of Registration Applications Reviewed and Submitted to the CFDA for Approval and Those Awaiting Approval in 2016 with Those from Three Years Prior
There is still a certain gap between China and developed countries in terms of the review time for imported drugs. The review time for new drugs in the United States is less than one year, while in Europe it is approximately one year. Since the initiation of reforms in the review process for domestically produced drugs, China has been continuously accumulating experience and enhancing its capabilities. Coupled with ongoing administrative reforms, the review times for both domestically produced and imported drugs are expected to be significantly shortened in the future.
What Is the Impact of the New Policy on Imported Drugs?
Returning to the new policy on imported drugs, if the reforms to clinical trials and marketing authorization applications for imported drugs prove effective, what magnitude of impact will they have on the domestic pharmaceutical market?
“China is a major producer of generic drugs, with nearly 5,000 pharmaceutical companies and more than 100,000 drug approval documents, the vast majority of which are for generics. The introduction of imported new drugs through the green channel will undoubtedly have a certain impact on the pharmaceutical market,” a healthcare sector analyst at a securities firm told VCBeat.
He believes that foreign countries possess superior capabilities in new drug development compared to China, whereas most domestic pharmaceutical companies are “sales-oriented,” prioritizing distribution channels and resources while underinvesting in R&D. “Among the top ten global pharmaceutical companies, even those primarily focused on generic drugs allocate more than 10% of their revenue to R&D. In contrast, very few Chinese companies exceed this threshold; for instance, a domestic pharmaceutical firm with annual sales nearing RMB 20 billion spends only 2% on R&D. This situation is bound to change,” stated the aforementioned securities analyst. He added that if multinational and domestic pharmaceutical companies were to compete on a level playing field, it would accelerate domestic firms’ strategic investments in R&D, thereby fostering positive developments in the overall landscape of new drug innovation.
“This process will not happen quickly, as domestic pharmaceutical companies still retain their existing channels and distribution networks. However, once foreign new drugs are approved for launch in the Chinese market, market forces will exert a reverse pressure, and pharmaceutical companies that fail to proactively innovate and compete will be eliminated by the market,” said the researcher.
However, multinational pharmaceutical companies are also selective about which drugs to prioritize in the Chinese market. First, they focus on treatments for major diseases with large domestic markets, including oncology, cardiovascular and cerebrovascular disorders, and antibody-based therapies. Second, they target drug categories where domestic technological capabilities are relatively weak, such as psychotropic agents and high-end biologics, allowing them to leverage first-mover advantages to capture market share and reap the benefits of original research and development.
Of course, multinational pharmaceutical companies and domestic manufacturers are not entirely in a competitive relationship; there is significant room for collaboration in clinical trials and joint research and development. Technological advantages and localization strengths will be key to future success. Currently, many pharmaceutical companies are actively pursuing overseas mergers and acquisitions to introduce new technologies, driven by the same rationale.
“In the context of a globalized market, competitive advantages built on ‘administrative barriers’ are not beneficial; more often than not, they come at the expense of local consumers’ interests. Taking the green channel for imported new drugs as an example, it is a positive development in the long run for both domestic pharmaceutical companies and patient populations. It places us on a level playing field with foreign pharmaceutical firms, enabling patients to access the benefits of new medications without delay,” Lu Sheng concluded.