Six Core Challenges Hindering Approval of Next-Gen Medical AI Products by China's NMPA
Editor’s Note: Retrospective data provide clinical support, the establishment of standardized databases, post-market clinical studies, technical black boxes, notifications of significant product changes, and differences between Chinese and U.S. regulatory approvals—all indicate that the approval pathway for China’s new generation of medical AI products remains arduous and long.Fortunately, the standard test databaseEstablishing, the standard dataset (fundus portion) was completed in just three months, and some companies will soon receive registration testing results issued by the National Institutes for Food and Drug Control (NIFDC). The construction of the standard database for pulmonary nodules has also been launched. See below for the specific standards obtained by VCBeat from enterprises.
In 2018, the medical artificial intelligence industry continued to experience rapid growth. In the first quarter, companies such as Huiyi Huiying, Infervision, Deepwise, Airdoc, and Shijian Medical all announced the completion of new rounds of financing, with amounts nearly all reaching the hundred-million-yuan level. United Imaging also launched its open intelligent AI platform, comprehensively penetrating the medical AI sector.
According to VCBeat (WeChat ID: vcbeat), although the industry is developing rapidly, one issue has consistently constrained its growth: no domestic company’s next-generation medical AI product has yet obtained a medical device registration certificate. This is partly because medical AI, as an entirely new category of products with certain intelligent capabilities, lacks prior regulatory experience and standardized databases for approval. On the other hand, medical AI products from Chinese companies are still in the refinement stage; healthcare is a highly rigorous industry that directly impacts public safety.
It is worth noting that most previously certified products were approved under the earlier CAD product approval framework, which differs somewhat from the newer generation of medical AI products that have recently gained prominence (this article focuses on the new generation of medical artificial intelligence products).
Without certification, there is no market access qualification. Although various startups have their own reasonable and legal revenue channels and substantial financing, AI companies face high costs for data, talent, computing power, and operations. If companies whose core business is medical AI products fail to obtain market access qualifications in a timely manner, their long-term development will inevitably face significant challenges.
Why Are Medical AI Products Developed with Deep Learning Technology Failing to Obtain Certification? Where Do the Problems Lie? What Issues Still Need to Be Resolved for Certification? What Preparations Have Various Medical AI Companies, Regulatory Agencies, and Other Institutions Made? What Are the Differences Between Approval Processes in China and the United States? VCBeat Attempts to Unravel These Mysteries.
1. Why Can't China Issue Certifications Like the United States?
In early April 2018, the FDA approved the software program for IDx-DR, the first autonomous artificial intelligence diagnostic device developed by IDx for use in primary care settings. It can diagnose diabetic retinopathy by analyzing retinal images without the involvement of specialized physicians.
VCBeat has learned that what appears to be a simple diagnosis took IDx a full 21 years to achieve. IDx is a company focused on developing autonomous clinical diagnostic algorithms; it spent seven years alone communicating with the FDA on how to evaluate the system and ensure its accuracy and safety.
Dr. Zhou Shaohua, Chief Expert in Medical Image Analysis at Siemens, once explained to reporters that the U.S. regulatory system is structured as follows.
The FDA regulates medical devices into three classes based on their intended use and the risk they pose to patients. Class I includes low-risk devices, such as medical gloves; Class II comprises moderate-risk devices, such as CT scanners; and Class III encompasses the highest-risk devices, such as stents.
There are two types of AI imaging systems: computer-aided detection (CADe) and computer-assisted diagnosis (CADx). CADe is used to detect abnormalities, while CADx assesses the presence of disease, such as its severity, classification, or prognosis.
The FDA has extensive experience in regulating CADe software and provides 510(k) guidance standards on how to conduct clinical performance evaluations. However, the FDA has historically classified CADx systems as Class III devices.
(510(k) submissions are premarket applications filed with the FDA to demonstrate that the device seeking market clearance is as safe and effective as a legally marketed device that is not subject to premarket approval (PMA), i.e., it is substantially equivalent. Applicants must compare the device seeking clearance to one or more similar devices currently on the U.S. market to derive and support the conclusion of substantial equivalence.)
However, according to VCBeat, most medical AI products that have currently received FDA approval are classified as Class II devices. In other words, they obtain approval by meeting lower regulatory thresholds. In contrast, China currently classifies next-generation medical AI as Class III medical devices, resulting in a different approval process.
2. It is recommended that companies consider certifying Class III medical devices
Guo Na, co-founder of Huiyi Huiying, stated that the AI products recently approved in the United States have all followed the Class II certification pathway, demonstrating safety and effectiveness through equivalence comparisons with traditional Clinical Decision Support Systems (CDSS). In contrast, regulations from China’s National Medical Products Administration (NMPA) are relatively stricter, with rigorous control over clinical evaluation pathways. Under current regulations, most domestic AI products must undergo clinical trials for evaluation, which is a time-consuming process. Currently, the NMPA has not signaled any relaxation of approval standards. According to the new classification catalog, auxiliary decision-making software that directly provides diagnostic and treatment recommendations is classified as Class III medical devices. If the software only provides quantitative values, such as bone mineral density, it is regulated as a Class II medical device. The majority of AI products on the market fall under Class III.
Yasen Technology informed VCBeat that although it has obtained a Class II medical device registration certificate after nearly two years of effort, the company is still preparing for the approval of a Class III certificate in accordance with the latest regulatory changes. On September 4, 2017, the China Food and Drug Administration (CFDA) released the new version of the "Medical Device Classification Catalog," in which the category of auxiliary diagnosis under medical software primarily addressed previously existing auxiliary diagnostic products, such as Computer-Aided Diagnosis (CAD) systems. However, this classification did not encompass automatic diagnostic systems, leaving the regulatory definition for the recently emerging artificial intelligence software products incomplete.
This classification catalog was reformulated by the relevant authorities based on prior experience and issues encountered in the classification, approval, and certification of auxiliary diagnostic products. However, there are no precedents for next-generation medical artificial intelligence products. Although some AI companies have currently submitted applications for certification to the National Medical Products Administration (NMPA), none has yet obtained approval for Class III medical devices.
VCBeat has learned from various companies that major players such as Yasen, Huiyi Huiying, Tumor Deepwise, Infervision, Shuimi, Airdoc, and Yitu Healthcare are actively submitting applications for Class III medical device registration. Yitu Healthcare stated that its entire product portfolio is undergoing Class III certification. Additionally, Airdoc submitted for testing China’s first server equipped with AI software pending regulatory approval.
3. A standard database for approval is being gradually established
One reason why medical AI products have not received approval is that the standard database used for approval is still under construction.
Data standards vary across regions and hospitals. Even if clinical trials conducted by a company at two major hospitals in Beijing yield perfect results, this does not guarantee that the product can be deployed in a county-level hospital; indeed, due to overfitting, it may fail to function in other healthcare settings. To verify the robustness (generalizability) of medical AI products, regulatory authorities are currently working on establishing standardized test databases.
Currently, the construction of standard test databases is carried out by disease type. According to information released on the official WeChat account of the National Institutes for Food and Drug Control (NIFDC),On March 26, 2018, the construction of the standard test dataset (fundus portion) was completed. The plan for building the standard database of pulmonary nodules has also been finalized, and its construction is expected to be completed in the near future.。
VCBeat has learned through industry consultations and statistical analysis that nine companies are currently participating in the construction of a standardized test database. Taking pulmonary imaging as an example, lung nodule image data will be collected from no fewer than five hospitals, with each company contributing 2,000 images. The image acquisition equipment must be from major manufacturers such as GE and Siemens, and the images must comply with the DICOM 3.0 protocol standard. Ethical approval from the Institutional Review Board (IRB) is required for data usage. A two-round, double-blind annotation process by attending physicians will be implemented.. The number of images is expected to exceed 10,000 cases.
A medical AI company engaged in research on diabetic retinopathy and participating in the construction of the National Institutes for Food and Drug Control (NIFDC) fundus database told VCBeat that it would soon receive the registration testing results issued by the NIFDC.
According to Liu Shiyuan, Director of the Department of Diagnostic and Interventional Radiology and Nuclear Medicine at Changzheng Hospital, Second Military Medical University, who participated in the construction of the standard test database, To ensure fairness and gain recognition from the majority of enterprises and institutions, the development of the standard test database adheres to the following three principles:
The first isGeneralizability, the data must be sourced from diverse hospitals across China and should not be limited to medical data from major cities such as Beijing, Shanghai, Guangzhou, and Shenzhen.
Secondly,Compatibility,Taking lung images as an example, the establishment of standard test databases currently accounts for CT images with varying slice thicknesses, including 5-mm, 1–2-mm, and even submillimeter images.
Third, the annotation of medical images must be standardized. Professor Liu Shiyuan stated that while collecting a certain volume of images is not difficult, the challenge lies in labeling the data. Physicians engaged in annotating standard test databases are recruited from those who have previously conducted medical AI research. After recruitment, these physicians are trained according to standardized annotation protocols before proceeding with the labeling process. The ultimate goal is to establish a standard testing database that is free from any corporate or algorithmic biases.。
4. Redefine Major Changes to Address Rapid Iteration Challenges
Medical AI products face a situation where product performance, algorithmic models, and application interfaces are rapidly updated. In this context, traditional approval processes for additions or upgrades clearly fail to meet the industry’s development needs.
If new certification rules are not introduced, the iteration cycle for medical imaging AI products would be 3–5 days under the traditional approval process, requiring companies to file reports with government agencies every week—a burden that would be unsustainable for both enterprises and regulators. In such a scenario, “cutting corners” would inevitably occur: although companies continuously update their systems, they fail to report these changes through formal channels, a practice that the industry is keen to avoid.
Suo Na, Deputy General Manager of Huaguang Innovation (Beijing) Technical Service Co., Ltd.Tell VCBeat that the current approval technical guidelines provide a clear example of version naming. Versions are generally designated by four codes: S, Y, Z, and B. “S” denotes major enhancements, “Y” minor enhancements, “Z” corrective updates and builds, and “B” minor bug fixes. Changes involving S, Y, or Z require registration of the change, whereas changes involving B do not. Companies can adopt this naming convention for their own versions to comply with regulatory requirements, thereby avoiding the burden of frequent change registrations.
We believe that the core of this issue lies in the definition of "major changes." We will continue to adhere to the traditional practice of filing reports for major changes, while standardizing what constitutes a major change.
First,Database Changes. If the product’s database undergoes a 10%–20% change (this figure remains uncertain until specific guidelines are issued), the company may file a report with the relevant authorities and submit a request for modification after the study results are available;
Second,Algorithm Changes. If the algorithmic framework changes, a change request can be submitted. Currently, many enterprises optimize and upgrade their algorithms based on open-source algorithms, and to ensure proper localization, they also incorporate peripheral systems such as pre-processing software. Since changes to algorithms are difficult to quantify, this provision is still under discussion.
5. The "black box" issue of artificial intelligence technology warrants further discussion
It is often heard in various contexts that deep learning technology is suspected of being a "black box," as the causal relationship between input and output results cannot be explained. According to the principles of scientific validation, the presence of such a technological black box makes the certification process relatively more difficult.
Currently, many companies in China utilize internationally available open-source technologies. Some leading Chinese artificial intelligence firms are capable of explaining the underlying principles, including the total number of layers and the specific functions of each layer. However, a significant portion of companies still lack this capability, which indirectly highlights the disparities among industry peers.
Sona stated that products lacking established review guidelines primarily rely on the individual expertise of lead reviewers and the experiential knowledge of expert committees convened by regulatory authorities to conduct technical assessments, with a primary focus on evaluating product safety and reliability.
We believe that while the regulatory authorities do not necessarily need to place excessive emphasis on this issue, companies should be well-prepared to address it. For traditional software systems, quality supervision departments do not require a step-by-step elucidation of every process; rather, they prioritize safety and efficacy. The purpose of the quality inspection department’s understanding of the underlying technical principles is to determine the appropriate black-box testing strategy. Therefore, companies must know how to respond to related inquiries.
6. Issues to Note Beyond Standard Databases
Currently, the development of most medical AI companies is stalled at the regulatory approval stage. The registration and approval process for medical software generally proceeds as follows:
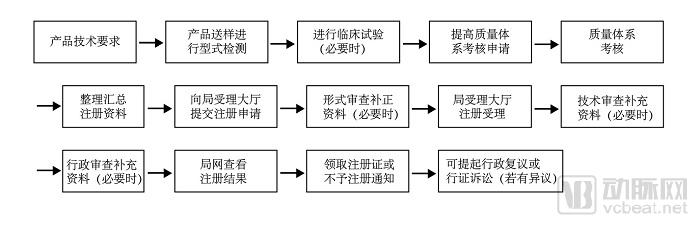
As shown in this flowchart, prior to obtaining certification from the National Medical Products Administration (NMPA), manufacturers must first secure quality management system certification, a process that takes 3–6 months. The certification for Class III medical devices requires 2–3 years, and approval is contingent upon the establishment of a standard database, which itself entails a considerable lead time.
Taking diabetic retinopathy as an example, on December 24, 2017, the official WeChat account of the National Institutes for Food and Drug Control (NIFDC) announced a notice on convening a meeting to develop AI standard test datasets (fundus portion). On March 26, 2018, the NIFDC’s official WeChat account announced the completion of the standard test dataset (fundus portion). The entire process for this single disease entity took only three months. Based on information released by the NIFDC via its official WeChat channel, it can be inferred that the database was constructed on a disease-specific basis. Compared with the seven years IDx spent communicating with the U.S. Food and Drug Administration (FDA) on how to evaluate systems and ensure their accuracy and safety, the approval authority’s pace was remarkably fast (notably including the Spring Festival holiday period).
However, each company typically focuses on around 10 disease indications. When accounting for the time required for clinical trials, the approval process takes approximately three to four years. This timeline is dictated by the inherent rigor and safety requirements of both healthcare and AI, representing an immutable reality for regulatory agencies and enterprises alike. Yet, how many companies can afford to wait this long? Should we consider a new approach to market access?
To ensure the sustainable development of enterprises and the industry, can we adopt the pharmaceutical industry’s model of “market launch first, clinical approval later,” provided that safety and stability are guaranteed, so as to foster initial industry growth?
(Broadly speaking, all clinical studies conducted after a drug is marketed are referred to as post-marketing studies. Narrowly defined, post-marketing clinical studies are application-related research on drugs independently organized by manufacturers, medical institutions, or social organizations.)
VCBeat has learned that, based on data collection methods, clinical studies can be categorized intoProspective Data Collection Studies and Retrospective Data Collection Studies. A study involving prospective data collection refers to one in which a research protocol is formulated in advance, and future data are collected in accordance with that protocol.A retrospective data collection study refers to the retrospective collection of historical data after the study protocol has been established.In other words, the primary difference between the two lies in whether the data collected are prospective or retrospective. The advantage of prospective data collection studies is that they allow for better control over data quality and yield more compelling evidence; however, their disadvantages include a longer research timeline and higher costs. In contrast, the advantages of retrospective data collection studies are a shorter research timeline and significantly lower costs.
Currently, we are adopting a prospective study approach. To encourage industry development, once certain products—such as AI-based medical devices for fundus imaging and pulmonary nodule detection—receive regulatory approval, other products may utilize retrospective studies to provide clinical support, thereby reducing the time and costs associated with clinical approval for enterprises.
Companies Should Engage More with Regulatory Authorities
The AI healthcare sector is an emerging industry, so it is inevitable that many regulations have yet to be established. Many companies have been in operation for only two to three years. Stakeholders should avoid short-sighted pursuits of quick success; healthcare is inherently a long-term industry. Both enterprises and regulatory bodies need time to gradually familiarize themselves with industry norms, as this is an unprecedented field.
During this process, enterprises should proactively engage with the quality control department. Both parties must communicate in depth to understand each other’s needs and develop a robust solution promptly, thereby facilitating administrative decision-making.