Spark Therapeutics: Pioneering the Future of Gene Therapy – IPO Prospectus Filed
By Gege
If human genes could be altered with little to no side effects, would this bring a new turning point to the future of humanity? It may sound like science fiction, but it is not. Consider Spark Therapeutics, a gene therapy company already approved by the U.S. FDA. “We don’t follow footsteps. We create the path.” May they truly spark the future of gene therapy.
Key Company: Spark Therapeutics (NASDAQ: ONCE)
Including the company: Shire (NASDAQ:SHPG)
Sangamo (NASDAQ:SGMO)
Nightstar Therapeutics(NASDAQ: NITE)
uniQure N.V.(NASDAQ: QURE)
Article Abstract
1. Spark Therapeutics, a pioneer in the field, holds the first globally approved gene therapy drug. The company was ranked among the Top 10 on MIT Technology Review’s “50 Smart Companies” list for two consecutive years (2016 and 2017) and was named one of Bloomberg Businessweek’s “50 Companies to Watch” in 2018.
2. On December 19, 2017, the first one-time gene therapy for retinal dystrophy caused by RPE65 gene mutations was approved by the U.S. FDA for marketing, marking a watershed moment in the field of gene therapy;
3. Philadelphia houses a 48,000-cubic-foot, state-of-the-art cGMP manufacturing facility, which is the first and only FDA-approved plant in the gene therapy sector;
4. The company has entered into an agreement with Novartis (NVS) for sales in Europe and other global markets excluding the United States. This is highly positive for an innovative technology company, laying a foundation for stable future revenue and long-term development. As of December 2017, the company held $540.2 million in cash; adding the $105 million upfront payment from the January collaboration with Novartis, the company currently maintains a robust cash position. Following de-risking, it stands as the preferred conservative-choice gene therapy company listed on the NASDAQ in 2018.

Spark Therapeutics is a gene therapy company that develops therapies for inherited retinal diseases (IRDs), neurodegenerative disorders, and other genetic conditions potentially curable through hepatic gene modification. This type of gene therapy typically offers a one-time treatment with the potential to cure the disease.
The company was founded in 2013 and listed on the NASDAQ in 2015. On December 19, 2017, its first one-time gene therapy drug for retinal dystrophy caused by RPE65 gene mutations was approved by the U.S. FDA for market launch, marking a watershed moment in the field of gene therapy. The company’s current market capitalization stands at $3 billion (as of the market close on April 20, 2018).
The company’s foundational technology originated from the Children’s Hospital of Philadelphia (CHOP). Its team of scientists, with over two decades of experience in the field of gene therapy, laid the groundwork for the subsequent development of the adeno-associated viral (AAV) vector platform, the establishment of manufacturing facilities, and research into the effects of AAV in bone, muscle, and liver tissues.
Spark Therapeutics employs more than 320 specialized professionals in the field and operates a 48,000-square-foot, state-of-the-art cGMP manufacturing facility in Philadelphia. This facility is the first and only one in the gene therapy sector to have received approval from the U.S. FDA. It produces adeno-associated virus (AAV) vectors in-house, with six production lines supporting clinical trials at more than 12 clinical trial sites, and performs final drug product assembly to meet clinical demand.
The company was ranked among the top 10 on MIT Technology Review’s “50 Smartest Companies” list for two consecutive years (2016 and 2017) and was named one of Bloomberg Businessweek’s “50 Companies to Watch” in 2018.
Pipeline Progress and Analysis
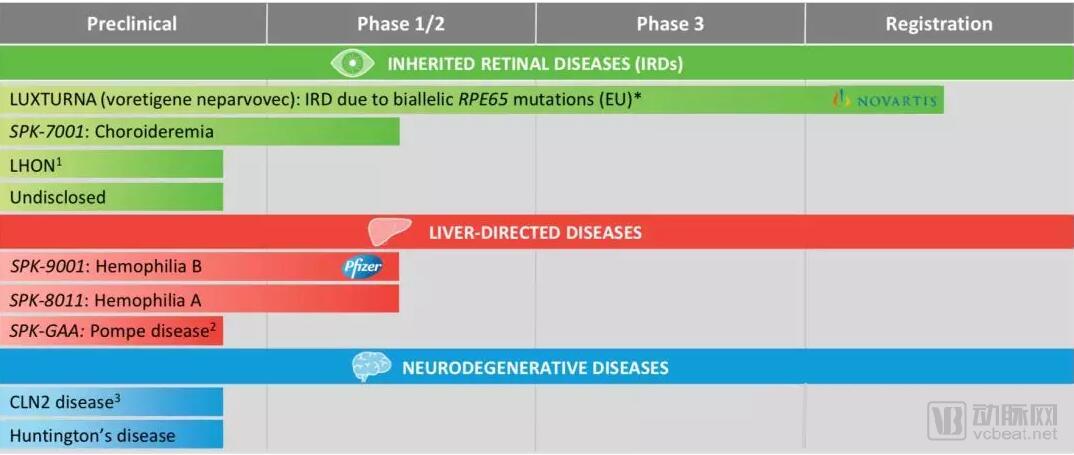
1. LUXTURNA(Voretigene neparvovec): biallelic RPE65-mediated IRD
Epidemiology, Market, and Pricing of RPE65 Mutation-Associated Inherited Retinal Diseases (IRDs)
Hereditary retinal diseases are a rare cause of blindness, resulting from mutations in more than 220 different genes. These conditions commonly affect children and young adults. Mutations in the RPE65 allele represent one such form. Patients inevitably progress to complete blindness, initially experiencing night blindness and involuntary nystagmus. As the disease progresses, patients first lose their peripheral vision, which develops into intermediate tunnel vision; ultimately, even the central visual field is lost, leading to total blindness.
Currently, there are no other approved drugs for inherited retinal dystrophy (IRD) caused by RPE65 mutations. According to epidemiological data estimated by Spark, there are approximately 6,000 patients with RPE65 mutations in the United States, Europe, and the Asia-Pacific region. In the United States, an estimated 1,000–2,000 patients suffer from blindness due to RPE65 mutations, with approximately 10–20 new cases diagnosed annually.
Post-listing,LUXTURNA is priced at $850,000 per treatment for both eyes.Spark stated that it would refund a portion of the cost to patients if LUXTURNA proved ineffective.
December 19, 2017LUXTURNAOfficially approved for marketing by the U.S. FDA, having previously received FDA Priority Review, Orphan Drug Designation, and Breakthrough Therapy Designation.
Following approval, the company also obtained a Priority Review Voucher for a rare pediatric disease, which currently has a market value of USD 100–200 million. Sales performance of this drug in the United States became visible in the first quarter of 2018. The New Drug Application (NDA) in Europe was submitted on July 31, 2017.
This drug previously received orphan drug designation in Europe, which grants up to 10 years of market exclusivity. The rights for Europe and global markets (excluding the United States) have been licensed to Novartis. The company received a $105 million upfront payment and $65 million in milestone payments, with future royalties amounting to approximately 25%.
LUXTURNA Phase 3 Clinical Trial Results
The results of the clinical trial were published in the prestigious journal *The Lancet*. Efficacy was determined based on changes in the one-year MLMT (Multi-Luminance Mobility Test) score. Due to retinal growth, this medication is contraindicated in patients under 12 months of age. Phase III clinical results demonstrated efficacy: among 21 treated patients and 10 controls, there were statistically significant differences after one year in the median bilateral MLMT score (P=0.001) and the median MLMT score for the first treated eye (P=0.003). Patients in the control group exhibited similar therapeutic benefits after crossing over to receive LUXTURNA. Regarding safety, adverse reactions included conjunctival hyperemia (22%), cataracts (20%), increased intraocular pressure (15%), retinal tears (10%), endophthalmitis (5%), and ocular pain (5%).
2. SPK-7001: Choroideremia(CHM)
Choroideremia (CHM) is an X-linked inherited retinal dystrophy (IRD) characterized by night blindness and visual field loss in adolescents, with progressive constriction of the visual field. CHM is caused by deletions or mutations in the CHM gene, leading to a deficiency of Rab escort protein 1 (REP-1). The company estimates that approximately 12,500 males in the United States and five European countries are affected by this disease.
This pipeline is in Phase 1/2 clinical trials, with 10 patients enrolled and divided into two dosage groups. Recently, the company has also recruited five patients at relatively earlier stages of the disease. Available data indicate that SPK-7001 is well tolerated, with no drug-related adverse events or serious adverse events (SAEs) observed.
As of March 29, 2017, five cases had completed one year of follow-up, four cases had completed 18 months of follow-up, and one case had nearly two years of follow-up. Interim data showed that four out of ten patients exhibited a difference in visual acuity between the eye receiving the investigational drug injection and the control eye. For safety assessment, these patients will be followed up for 15 years.SPK-7001 has received Orphan Drug Designation in the United States and Europe.
RPE65 and CHM are two of the more than 220 gene mutations associated with inherited retinal diseases (IRDs). ONCE is also conducting corresponding preclinical studies on other IRDs, such as Leber hereditary optic neuropathy.
3. SPK-9001: Hemophilia B
Hemophilia B is a coagulation disorder caused by a deficiency of functional factor IX (FIX), a clotting factor produced in hepatocytes. Mutations in the gene encoding the FIX protein in liver cells lead to the development of this disease. The condition is characterized by recurrent, life-threatening spontaneous bleeding episodes. According to the 2016 World Federation of Hemophilia Annual Global Survey, there are approximately 30,000 patients worldwide.
Severe hemophilia B is defined as a factor IX (FIX) level of less than 1% of normal. Patients with this condition frequently experience spontaneous bleeding, leading to hemorrhage into joints and muscles. Moderate hemophilia B is defined as a FIX level between 1% and 5% of normal. Current treatment primarily involves prophylaxis and intravenous infusion of factor IX.
In 2014, the Company entered into an agreement with Pfizer (NYSE: PFE), whereby, upon completion of the Phase 1/2 clinical trials of SPK-9001 by the Company, Pfizer would assume responsibility for subsequent research and development, regulatory approval applications, and commercialization.2016, received FDA Breakthrough Therapy Designation.
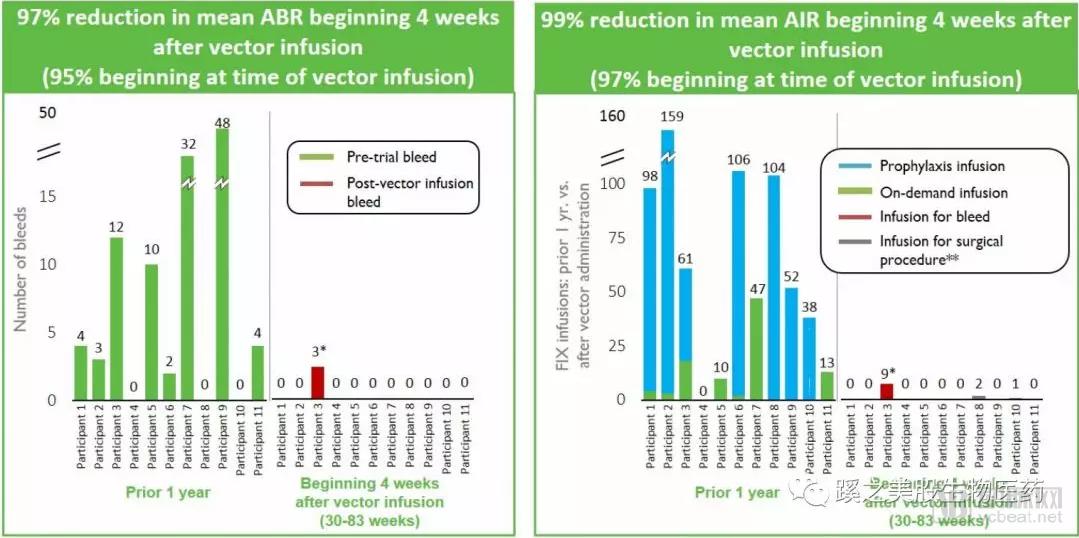
Clinical Trial Results: Efficacy:As of November 29, 2017, the first cohort of 11 patients received a dose of 5 × 10¹¹ vector genomes per kilogram of body weight. All 11 patients discontinued Factor IX infusions, and their circulating Factor IX levels remained stable. Compared with pre-enrollment data, the overall annualized bleeding rate (ABR) decreased by 97%, and the overall annualized infusion rate (AIR) decreased by 99%.
Factor IX activity in the blood remained at 36% of normal levels (range as of the data cutoff: 15–78%) 12 weeks after dosing. The company will also enroll four patients in a Phase 1/2 clinical trial to receive a booster regimen of SPK-9001, aiming to assess the compatibility of the new booster treatment with SPK-9001.
Safety: No serious adverse events, thrombotic events, or development of Factor IX inhibitors (antibody formation) were observed. Two patients experienced asymptomatic, transient elevations in liver enzymes, which were resolved with oral corticosteroids. One patient with severe joint disease received coagulation factor infusion due to suspected hemarthrosis.
4. SPK-8011: Hemophilia A
Hemophilia A is caused by gene mutations in liver cells, leading to a deficiency in factor VIII production and resulting in recurrent spontaneous bleeding. Similar to hemophilia B, it is classified as severe hemophilia A when clotting factor levels are below 1%, and as moderate hemophilia A when levels range from 1% to 5%. The clinical manifestations are largely similar to those of hemophilia B. According to the 2016 World Federation of Hemophilia Annual Global Survey, there are approximately 150,000 patients worldwide. In February 2018, it receivedFDA's Breakthrough Therapy DesignationThe Company reserves the rights for global commercialization.
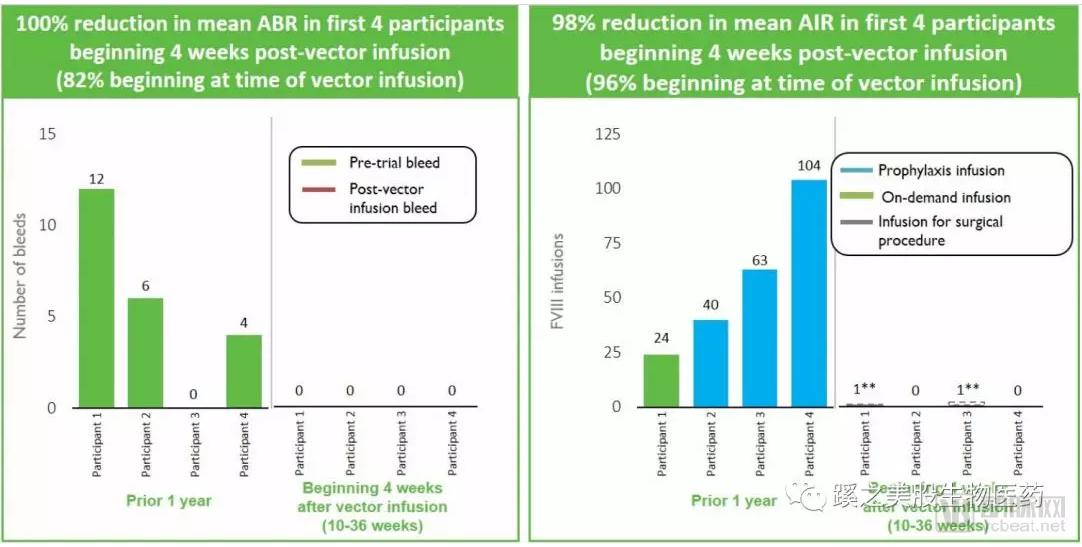
Clinical Trial Results: Efficacy: As of December 6, 2017, the first cohort of four patients received doses of 5 × 10¹¹ or 5 × 10¹² vector genomes per kilogram of body weight. The overall annualized bleeding rate (ABR) was reduced by 100%, and the overall annualized infusion rate (AIR) was reduced by 98%. Safety: No serious adverse events, thrombotic events, or development of Factor VIII inhibitors were observed.
Comment: The market for hemophilia A is substantial, with competitors including Shire (NASDAQ:SHPG) and Sangamo (NASDAQ:SGMO). The company’s pipeline involves a small patient population, and there is wide variability in Factor VIII levels in the blood after 12 weeks, suggesting that dose exploration still has a long way to go. While some argue that the company’s drug candidate for this indication is ineffective, I believe this likelihood is very low. Competitor BMRN has reported significantly better data, so close monitoring will be necessary in the later stages.
5. CNS-directed gene therapies
For two neurodegenerative diseases, Batten disease and Huntington's disease, the company is primarily developing therapies targeting patients with TPP1 deficiency to treat these conditions. Both pipelines are currently in preclinical trials. Both pipelines have obtainedOrphan Drug Application.
In summary, Spark Therapeutics has the first approved drug in global gene therapy and is a pioneer in this field. It operates the world’s most advanced cGMP manufacturing facility in Philadelphia, which is also the first and only gene therapy production facility approved by the U.S. FDA.
The company was ranked among the top 10 on MIT Technology Review’s “50 Smartest Companies” list for two consecutive years (2016 and 2017) and was also named one of Bloomberg Businessweek’s “50 Companies to Watch” in 2018.
The sales agreement for Europe and the rest of the world (excluding the United States) signed with Novartis (NVS) represents a highly positive partnership for this innovative technology company, laying the foundation for stable future revenue and long-term development.
The company currently has ample cash reserves and low risk, with a market capitalization of $3 billion, making it the top choice for a stable NASDAQ-listed gene therapy company in 2018.
Market Competitors
The market for hemophilia A is enormous, with competitors including Shire (NASDAQ:SHPG) and Sangamo (NASDAQ:SGMO), among others;
Competitors in the field of choroideremia include Nightstar Therapeutics (NASDAQ: NITE);
Hemophilia B competitors include uniQure N.V. (NASDAQ: QURE);
Valuation (Cash and Valuation)
As of December 2017, the Company had cash reserves of $540.2 million. Coupled with the $105 million upfront payment from its collaboration with Novartis in January, the Company currently maintains a robust cash position. In August 2017, the Company conducted an initial public offering (IPO), raising a total of $380.4 million. The underwriters exercised all options, at an offer price of $76.00 per share.
Institutional and Management Holdings
The company’s management team is primarily composed of alumni from the Children’s Hospital of Philadelphia. Under the leadership of CEO Jeff Marrazzo, the company has raised $1 billion in financing over four years, built a team of 320 employees, and received numerous awards. Truly outstanding!
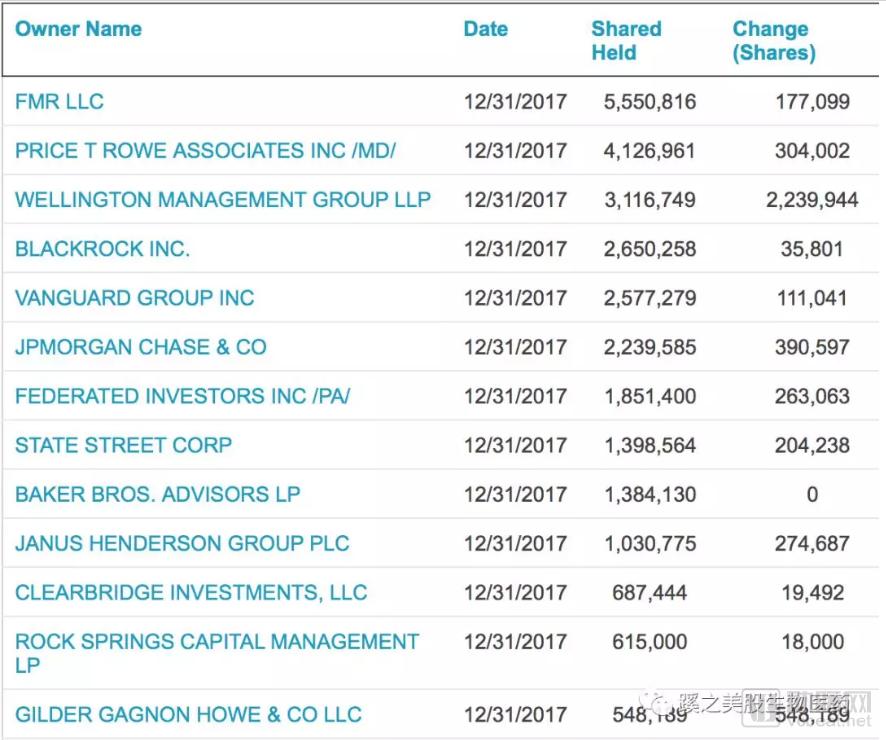
The top ten shareholders included prominent biotech investment firms such as Baker Bros., and institutional investors significantly increased their holdings in the fourth quarter of 2017.
Investment Risks
The company’s approved products have seen slower-than-expected market uptake, and their safety profile requires long-term observation over a 15-year period to monitor for any additional adverse reactions, which remains an uncertain factor. Meanwhile, competitors are advancing R&D at a faster pace with superior outcomes, and new entrants are joining the competitive landscape.
Author: Gege
US Biopharmaceutical Research Analyst