2026 therapy pipeline to watch: 10 promising candidates reshaping treatment paradigms
At the beginning of 2026, the latest report from Evaluate highlights ten innovative therapies, spanning multiple fields such as metabolic diseases, oncology, and autoimmune disorders, that it projects are poised for potential FDA approval this year. These therapies not only exemplify scientific progress but also herald shifts in treatment paradigms in the coming years.
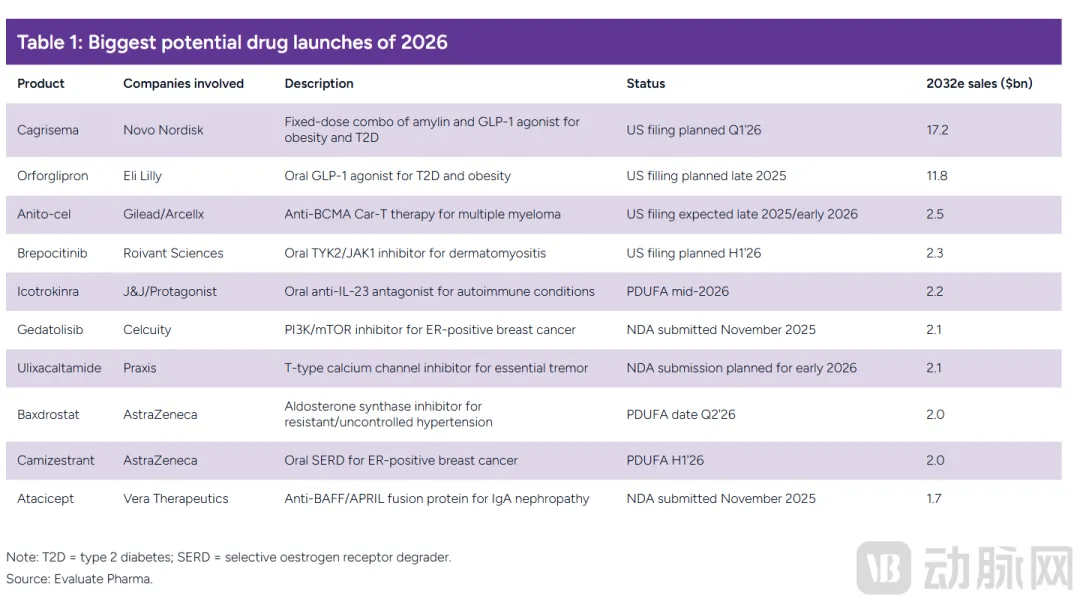
The Most Promising Pre-Commercial Drugs of 2026 (Source: Evaluate)
01
"Dual Giants Compete" in the Metabolic Disease Arena: New Narratives Beyond GLP-1
In the field of metabolic disease treatment, particularly the obesity therapeutics market, the competition between the two giants, Novo Nordisk and Eli Lilly, has consistently garnered significant attention. In 2026, with the submission of New Drug Applications for Novo Nordisk's CagriSema and Eli Lilly's orforglipron, competition in this space is intensifying, potentially reshaping the obesity treatment landscape.
CagriSema is a fixed-dose combination therapy consisting of the long-acting amylin analog cagrilintide (2.4 mg) and semaglutide (2.4 mg), designed for once-weekly subcutaneous injection. Both amylin and GLP-1 are hormones secreted by the gut after meals, and they possess synergistic mechanisms of action. Amylin slows gastric emptying, inhibits glucagon secretion, thereby reducing postprandial glucose fluctuations, and also acts on the central nervous system to increase satiety. Semaglutide, as a GLP-1 receptor agonist, stimulates insulin secretion, suppresses appetite, and reduces food intake. By combining the two, CagriSema aims to regulate metabolism more effectively through multiple pathways to achieve weight loss.
Eli Lilly's orforglipron, on the other hand, is an investigational, once-daily oral small-molecule glucagon-like peptide-1 (GLP-1) receptor agonist that can be taken at any time of day without restrictions related to food or water intake. It binds to the GLP-1 receptor, activating downstream signaling pathways to exert physiological effects similar to native GLP-1, such as promoting insulin secretion, inhibiting glucagon release, and delaying gastric emptying, thereby aiding in glycemic control and weight reduction. Its small-molecule structure confers inherent advantages for oral absorption, bypassing the delivery hurdles common to peptide-based drugs.
Regarding the mode of administration, CagriSema's once-weekly subcutaneous injection may pose issues of inconvenience or needle phobia for some patients. In contrast, orforglipron's once-daily oral administration undoubtedly greatly improves patient compliance and better aligns with modern patients' demand for convenient treatment.
In terms of clinical data, the NDA for CagriSema is primarily based on results from two Phase 3 clinical trials, REDEFINE 1 and REDEFINE 2. Results from the REDEFINE 1 trial showed that, regardless of whether patients continued treatment, the CagriSema group achieved a 20.4% reduction in body weight at week 68, compared to 3.0% for the placebo group, a statistically significant difference. Under the assumption that all patients continued treatment, the weight reduction in the CagriSema group at week 68 was 22.7%, compared to 2.3% for the placebo group. Furthermore, 91.9% of participants in the CagriSema group achieved weight loss of 5% or more, compared to 31.5% in the placebo group.
For orforglipron, results from the Phase 3 ATTAIN-2 trial, announced last August, showed that for adults with obesity or overweight and type 2 diabetes, all three doses of orforglipron met the primary and all key secondary endpoints. Over 72 weeks of treatment, the drug led to significant weight loss, meaningful reductions in HbA1c levels, and improvements in cardiometabolic risk factors. Positive topline results from the Phase 3 ATTAIN-MAINTAIN trial, announced in December, indicated that orforglipron also met the primary and key secondary endpoints for maintaining weight loss.
Regarding the magnitude of weight loss, CagriSema's results under the optimal assumption show a slightly greater reduction than the published results for orforglipron. However, orforglipron also demonstrates excellent performance in glycemic control and improving cardiometabolic risk factors, and its convenient oral administration may attract more patients.
These two drugs represent two major trends in obesity treatment: combination therapies and oral GLP-1 agonists. Combination therapies, by integrating multiple mechanisms of action, promise more comprehensive and effective treatment outcomes. Oral GLP-1 agonists address the compliance issues associated with traditional injectable formulations. With the potential approval and market launch of these two drugs in 2026, the obesity treatment market is set to enter a new competitive landscape, offering patients more and better treatment options. This will not only drive progress in the field of obesity treatment but may also have a profound impact on the broader metabolic disease therapeutics market, encouraging more pharmaceutical companies to increase R&D investment in this area and explore further innovative therapies.
02
Further Evolution of Cell Therapy: How Can BCMA CAR-T Break the Deadlock in Solid Tumors?
As an emerging force in the field of cancer treatment, cell therapy has made significant progress in recent years. Chimeric Antigen Receptor T-cell (CAR-T) therapy, in particular, has demonstrated remarkable efficacy in treating hematological malignancies. However, extending the success of CAR-T therapy to solid tumors remains a major challenge for researchers and clinicians. In 2026, anitocabtagene autoleucel (anito-cel), co-developed by Gilead Sciences and Arcellx, may offer a new potential breakthrough direction for this challenge.
Anitocabtagene autoleucel is a CAR-T cell therapy targeting B-cell maturation antigen (BCMA). Its unique feature lies in the innovative, compact binding domain developed by Arcellx—the D-Domain. Traditional CAR-T cell therapies often face issues such as severe immune toxicity and unstable CAR expression during treatment.
The design of the D-Domain effectively addresses these challenges. It is small and stable, enabling high CAR expression without inducing persistent signaling. This characteristic allows CAR-T cells to maintain high activity in recognizing and eliminating tumor cells while avoiding severe toxic reactions caused by over-activation of the immune system. Furthermore, the D-Domain is designed to disengage rapidly from the BCMA target, further reducing the risk of immune-related adverse events, thereby ensuring safety while effectively clearing multiple myeloma cells.
In the treatment of multiple myeloma, anitocabtagene autoleucel has shown excellent efficacy. Key Phase 2 clinical trial data from the iMMagine-1 study, presented at the American Society of Hematology Annual Meeting last December, revealed that at a median follow-up of 15.9 months, patients with relapsed/refractory multiple myeloma treated with anito-cel achieved an overall response rate of 96% and a complete response/strict complete response rate of 74%. The 24-month progression-free survival rate was 61.7%, and the 24-month overall survival rate was 83.0%. These data indicate that anitocabtagene autoleucel offers an effective and durable treatment option for patients with multiple myeloma, significantly improving their survival outcomes.
Examining the advancement of CAR-T in multiple myeloma treatment, from initial exploratory applications to continuous optimization and upgrades, each technological innovation has brought better therapeutic efficacy and safety. Building upon first-generation CAR-T, subsequent developments introduced second-generation CAR-T with co-stimulatory domains, enhancing T-cell activity and persistence, while third-generation CAR-T added further co-stimulatory domains aiming for stronger immune activation. The emergence of anitocabtagene autoleucel represents innovation at the structural design level, addressing key issues faced by traditional CAR-T through the D-Domain and propelling CAR-T therapy to new heights in multiple myeloma treatment.
Regarding the potential future expansion into solid tumors, the success of anitocabtagene autoleucel provides valuable experience for the development of other CAR-T therapies for solid tumors. Solid tumors have been a difficult area for CAR-T treatment due to their complex tumor microenvironment, heterogeneity, and lack of specific tumor antigens. However, the advantages demonstrated by the D-Domain—such as high expression, low immune toxicity, and rapid disengagement—may be applied to solid tumor treatment through rational target selection and structural optimization.
For example, designing D-Domain-based CAR-T cells targeting certain solid tumor-specific antigens holds promise for overcoming the suppressive tumor microenvironment and achieving effective killing of solid tumor cells. Although extending CAR-T therapy to solid tumors still faces numerous scientific challenges—including improving CAR-T cell infiltration efficiency into solid tumor tissues and overcoming immune-suppressive signals within the tumor microenvironment—the research, development, and clinical application of anitocabtagene autoleucel offer new research directions and technical insights for breakthroughs in this field.
03
Autoimmune Diseases: From "Suppressing Inflammation" to "Precision Blockade"
Autoimmune diseases are chronic, progressive, and difficult-to-treat disorders caused by abnormal recognition of self-tissues by the immune system, leading to damage. Traditional treatments primarily rely on non-specific immunosuppressants such as corticosteroids. While these can alleviate inflammation, their lack of targeting often impairs normal immune function and triggers various adverse effects.
Research into the pathogenesis of autoimmune diseases has confirmed that abnormalities in specific cytokines and dysregulation of signaling pathways are core drivers of disease. This understanding has shifted therapeutic development from broad-spectrum suppression toward precise targeting. Brepocitinib and icotrokinra, which are anticipated to gain approval in 2026, provide crucial support for the optimization of precision therapeutic strategies in this field and represent the forward direction of its development.
Brepocitinib is a selective TYK2/JAK1 dual inhibitor. TYK2 and JAK1 are key nodes in cytokine signal transduction, mediating the signaling of pro-inflammatory cytokines such as Type I interferons, IL-6, and IL-12/IL-23, which are central drivers of autoimmune diseases and can trigger aberrant immune activation and damage to self-tissues. By specifically inhibiting the activity of TYK2/JAK1, brepocitinib precisely blocks the pathogenic signaling pathways, avoiding broad suppression of normal immune function and thereby achieving enhanced efficacy with reduced toxicity.
Data from the pivotal Phase III VALOR clinical trial, announced in September 2024, showed that brepocitinib met the pre-specified primary and key secondary endpoints in patients with dermatomyositis (DM). Primary endpoint analysis revealed that at week 52, the mean Total Improvement Score (TIS) in the brepocitinib group was 46.5, significantly higher than the 31.2 in the placebo group (p=0.0006), indicating that the drug can significantly improve clinical symptoms and thereby enhance the quality of life for DM patients. Subgroup analysis showed that the proportion of patients in the brepocitinib group who discontinued background corticosteroid treatment was nearly double that in the placebo group, suggesting its potential to reduce patients' reliance on traditional medications like corticosteroids and further lower the risk of adverse effects associated with long-term steroid use. This offers a superior treatment option for patients with dermatomyositis.
Icotrokinra is a potential first-in-class oral targeted peptide whose core mechanism of action is the specific blockade of the interleukin-23 receptor (IL-23R). IL-23 is a key pro-inflammatory factor in the pathological microenvironment of autoimmune diseases. It plays an indispensable regulatory role in the activation of pathogenic T cells in conditions such as moderate-to-severe plaque psoriasis and is a central molecule mediating chronic inflammation in these diseases.
Icotrokinra specifically binds to IL-23R, competitively blocking the interaction between IL-23 and its receptor, thereby inhibiting the activation of downstream inflammatory signaling pathways and curbing the initiation and progression of pathological inflammation at its source. This precise targeting of IL-23R enables specific treatment for related autoimmune diseases like psoriasis, avoiding non-specific interference with the body's normal immune function.
Clinical studies conducted jointly by Johnson & Johnson and Protagonist Therapeutics have confirmed the therapeutic efficacy of icotrokinra. The drug met both the primary and co-primary endpoints in four Phase III clinical trials for psoriasis. Topline data from the pivotal Phase III ICONIC-LEAD trial, announced in April 2025, demonstrated that once-daily oral icotrokinra significantly improved skin lesions in patients with moderate-to-severe plaque psoriasis with a favorable safety profile.
At week 16, 65% of patients in the icotrokinra group achieved an Investigator's Global Assessment (IGA) score of 0 or 1 (clear or almost clear skin), and 50% achieved a Psoriasis Area and Severity Index (PASI) 90 response (≥90% symptom improvement), compared to only 8% and 4%, respectively, in the placebo group. By week 24, response rates further improved: 74% of patients achieved IGA 0/1, of which 46% achieved IGA 0 (complete clearance), 65% achieved PASI 90, and 40% achieved PASI 100 (complete response). These clinical data robustly validate the efficacy and safety of icotrokinra in treating psoriasis, providing a new therapeutic option for patients with moderate-to-severe plaque psoriasis.
Beyond therapeutic efficacy, the mode of administration for both brepocitinib and icotrokinra holds significant clinical importance for improving patient adherence. Autoimmune diseases often follow a chronic, progressive course requiring long-term maintenance therapy, and patient adherence directly impacts clinical outcomes. Among traditional treatments, injectable formulations and high-frequency dosing regimens can increase the treatment burden, leading to reduced adherence and consequently affecting therapeutic effectiveness.
Both brepocitinib and icotrokinra are administered orally. Icotrokinra, in particular, is dosed once daily, simplifying the treatment regimen and reducing the complexity for patients. The oral route avoids the trauma and discomfort associated with injections, allows for convenient at-home self-administration, enhances treatment convenience, and can significantly improve long-term adherence. This helps ensure the standardized implementation of the treatment plan, ultimately optimizing patients' quality of life and facilitating their return to normal social activities.
The development and clinical application of brepocitinib and icotrokinra mark an important breakthrough in the field of autoimmune disease treatment, shifting from broad inflammation suppression to precise mechanistic blockade. By precisely targeting key disease signaling pathways, they demonstrate significant advantages in clinical efficacy. Furthermore, their oral administration modes enhance patient treatment adherence, underscoring their substantial clinical value. If successfully approved for market launch in 2026, they will provide patients with autoimmune diseases more effective, safer, and more convenient treatment options, driving paradigm shifts and advancing the discipline of autoimmune disease management.
04
Has Breast Cancer Treatment Entered the "Combination Era"? Could the PAM Pathway Be the Key to a Breakthrough?
HR+/HER2- advanced breast cancer accounts for 70% of all breast cancer patients. While dependent on endocrine therapy, drug resistance poses a significant challenge, severely limiting patient prognosis. Traditional treatment has primarily relied on single-agent endocrine therapies such as tamoxifen. However, 30-40% of patients exhibit primary resistance, and among those initially responsive, secondary resistance often develops within 5-12 months. Treatment options post-resistance are limited, with chemotherapy carrying significant toxicity and side effects that severely impact quality of life.
The PAM pathway plays a crucial role in the occurrence, progression, and drug resistance of HR+/HER2- advanced breast cancer. In normal cells, this pathway regulates physiological processes such as growth and proliferation. However, over half of patients harbor genetic alterations related to this pathway (e.g., activating AKT1 mutations, PTEN loss/mutation), leading to dysregulation of the pathway, which not only promotes tumor progression but also induces resistance to endocrine therapy.
Gedatolisib is an investigational multi-target PAM inhibitor that potently targets all Class I PI3K isoforms as well as mTORC1/2, achieving comprehensive pathway blockade. Unlike single-target inhibitors, which can easily trigger cross-activation and have limited efficacy, its comprehensive inhibition reduces adaptive cross-activation, sufficiently blocks the pathway, and restores tumor cell sensitivity to treatment.
Celcuity's related research has demonstrated the exceptional efficacy of gedatolisib. In November of last year, Celcuity announced the completion of its New Drug Application (NDA) submission to the U.S. FDA for gedatolisib, intended for the treatment of patients with HR-positive, HER2-negative advanced breast cancer. The NDA submission was primarily based on results from the PIK3CA wild-type patient cohort in the Phase 3 VIKTORIA-1 clinical trial. This trial evaluated the efficacy and safety of the gedatolisib combination in patients with HR-positive, HER2-negative advanced breast cancer whose disease had progressed after treatment with a CDK4/6 inhibitor and an aromatase inhibitor.
Results from the analysis of the PIK3CA wild-type subgroup showed that, compared to the active comparator, the triple regimen of gedatolisib, palbociclib, and fulvestrant reduced the risk of disease progression or death by 76%. The median progression-free survival (PFS) reached 9.3 months, compared to just 2.0 months in the comparator arm, representing an extension of 7.3 months (HR=0.24, 95% CI: 0.17-0.35, p<0.0001). The objective response rate (ORR) was 31.5%, with a median duration of response (DOR) of 17.5 months. These data fully demonstrate that the combination of gedatolisib with other drugs can significantly improve treatment outcomes and extend progression-free survival for HR+/HER2- advanced breast cancer patients with drug resistance.
In addition to gedatolisib, the next-generation oral selective estrogen receptor degrader (SERD) camizestrant also brings new hope for the treatment of HR+/HER2- advanced breast cancer. Camizestrant, developed by AstraZeneca, is a novel agent that not only blocks estrogen receptor (ER) signaling but also further induces ER degradation, fundamentally cutting off the growth signals for breast cancer cells. Compared to traditional endocrine therapies, camizestrant features an optimized molecular structure that significantly enhances its ability to degrade mutant ER, showing particularly better efficacy in patients with ESR1 mutations. In HR+/HER2- advanced breast cancer patients, ESR1 mutation is a major cause of endocrine therapy resistance, present in approximately 30% of advanced patients.
Multiple clinical trials conducted by AstraZeneca have confirmed the efficacy of camizestrant. In the Phase 2 SERENA-2 study, PFS was significantly longer in the camizestrant group compared to the fulvestrant group (7.2 vs. 3.7 months, HR=0.59). More critically, in the ESR1 mutation subgroup, patients in the camizestrant group achieved an even more pronounced PFS benefit (6.3 vs. 2.2 months, HR=0.33). An analysis of the Phase 3 SERENA-6 trial, announced last June, showed that the combination of camizestrant with CDK4/6 inhibitors (palbociclib, ribociclib, or abemaciclib) resulted in a highly statistically significant and clinically meaningful improvement in progression-free survival.
The SERENA-6 trial evaluated the efficacy and safety of switching from the standard first-line treatment (aromatase inhibitor combined with a CDK4/6 inhibitor) to a combination regimen including camizestrant in HR-positive, HER2-negative advanced breast cancer patients harboring ESR1 mutations. Results showed that, compared to the standard treatment regimen, the camizestrant-containing combination regimen reduced the risk of disease progression or death by 56% (HR=0.44; 95% CI: 0.31–0.60; p<0.00001). The PFS for patients switched to the camizestrant combination was 16.0 months, compared to 9.2 months in the control group. Notably, consistent PFS benefits were observed across all clinically relevant subgroups, regardless of the type of CDK4/6 inhibitor used.
The emergence of gedatolisib and camizestrant marks a significant shift in HR+/HER2- advanced breast cancer treatment from single-target to multi-pathway combination therapy. This combination approach is no longer confined to a single mechanism of action but instead exerts synergistic antitumor effects by simultaneously targeting multiple key nodes and signaling pathways. On one hand, the comprehensive inhibition of the PAM pathway by gedatolisib can effectively overcome resistance caused by abnormal pathway activation and restore tumor cell sensitivity to treatment. On the other hand, camizestrant blocks and degrades the estrogen receptor, inhibiting tumor cell growth at its root. The combination of the two attacks tumor cells from different angles, providing a more comprehensive and effective treatment strategy for patients with HR+/HER2- advanced breast cancer.
For patients with drug resistance, this multi-pathway combination therapy model holds significant breakthrough value. In the past, treatment options for HR+/HER2- advanced breast cancer patients after resistance were extremely limited, often leading to poor prognosis. Now, the combined application of gedatolisib and camizestrant opens new therapeutic avenues for these patients. By using methods such as genetic testing to identify patients with PAM pathway abnormalities and ESR1 mutations, and administering the targeted combination regimen accordingly, patients' progression-free survival can be significantly extended, and their quality of life improved. Furthermore, this combination therapy model provides new directions and insights for the future development of breast cancer treatment, encouraging more pharmaceutical companies to increase investment in R&D within the field of multi-pathway combination therapy and promoting continuous advancements in breast cancer treatment technology.
05
Hypertension, Tremor, IgA Nephropathy... Those Overlooked "Niche Battlefields"
In the biopharmaceutical field, certain niche diseases with low incidence or nonspecific symptoms, though easily overlooked, cause significant distress to patients, with treatment needs remaining unmet for a long time. In 2026, several innovative therapies targeting such conditions are expected to gain approval, offering hope to patients and reflecting pharmaceutical companies' strategic focus and exploration in areas of high unmet need.
Hypertension is a common global disease, with WHO 2025 data showing approximately 1.4 billion patients worldwide. Uncontrolled hypertension—where blood pressure remains elevated despite treatment with two or more antihypertensive drugs—is a major clinical challenge. Long-term uncontrolled hypertension significantly increases the risk of myocardial infarction, stroke, and renal failure. Although various traditional antihypertensive drugs exist, a substantial number of patients still fail to achieve adequate blood pressure control.
AstraZeneca's baxdrostat offers a new treatment option for these patients. It is a once-daily, highly selective, oral aldosterone synthase inhibitor (ASI) with the potential to become a first-in-class therapy. Its mechanism of action involves specifically inhibiting aldosterone synthase, the enzyme responsible for aldosterone synthesis in the adrenal glands. Aldosterone is a hormone that increases blood pressure by promoting sodium and water retention, and about 25% of hypertension cases are associated with dysregulated aldosterone levels. By blocking aldosterone production, baxdrostat reduces sodium and water retention, thereby lowering blood pressure.
In the Phase 3 BaxHTN trial, 796 adults with uncontrolled hypertension (on maximum tolerated doses of drugs from two different classes, including at least one diuretic) or resistant hypertension (on ≥3 different classes of maximum tolerated antihypertensive drugs, including at least one diuretic) were enrolled. Participants were randomized 1:1:1 to receive baxdrostat 2mg, baxdrostat 1mg, or placebo once daily in addition to their existing standard of care. The primary endpoint was the change from baseline in mean seated systolic blood pressure (SBP) at week 12.
Results showed that both doses of baxdrostat demonstrated a statistically significant and clinically meaningful reduction in mean SBP at week 12 compared to placebo. Improvements in secondary efficacy endpoints were also observed in the baxdrostat groups compared to placebo, including seated SBP in the resistant hypertension subgroup, seated diastolic blood pressure (DBP), and the proportion of patients achieving seated SBP <130 mmHg. Baxdrostat was generally well-tolerated in the trial.
Furthermore, positive full results from the Phase 3 Bax24 trial, announced on November 11, showed that compared to placebo, baxdrostat achieved a statistically and clinically significant reduction in 24-hour mean systolic blood pressure at week 12. Patients with resistant hypertension (rHTN) received baxdrostat 2mg or placebo on top of standard therapy, with the efficacy observed throughout the 24-hour cycle, including the morning period when cardiovascular event risk is higher in hypertensive patients.
Baxdrostat also demonstrated statistically and clinically significant reductions in multiple key secondary endpoints, including nocturnal ambulatory mean SBP (placebo-adjusted reduction of 13.9 mmHg) and seated SBP (placebo-adjusted reduction of 10.3 mmHg). A significantly higher proportion of patients treated with baxdrostat (71%) achieved a 24-hour mean SBP <130 mmHg compared to those receiving placebo (17%). These data fully demonstrate that baxdrostat, with its long half-life and high selectivity for aldosterone synthase inhibition, can significantly improve 24-hour and nocturnal blood pressure in patients with resistant hypertension.
Essential tremor is the most common movement disorder, characterized by involuntary, rhythmic shaking of the hands, with or without tremor in other body parts such as the head, voice, or legs. These tremors severely disrupt daily life and are progressive, typically increasing in severity and amplitude over time. In the United States, the beta-blocker propranolol is the only drug approved for essential tremor, but its efficacy is limited, tolerability is poor, and it is contraindicated in comorbidities affecting a significant portion of patients with essential tremor. Other beta-blockers and anticonvulsants are used off-label but similarly offer limited efficacy and tolerability.
Ulixacaltamide is a highly selective, small-molecule T-type calcium channel inhibitor that alleviates tremor by blocking abnormal neuronal firing in the cerebello-thalamo-cortical (CTC) circuit. Its efficacy and safety were validated in the Phase 3 Essential program, which comprised two concurrent trials. Study 1, a 1:1 randomized, double-blind, placebo-controlled, 12-week study, showed statistically significant improvements from baseline in the mADL11 score at week 8 for the ulixacaltamide group (-4.3 vs. -1.7 for placebo, p<0.0001) and across all key secondary endpoints. Study 2, a randomized withdrawal trial, showed that a significantly higher proportion of patients maintained efficacy in the ulixacaltamide group (55%) compared to the placebo group (33%, p=0.037). The drug was well-tolerated, with common adverse events including constipation and dizziness. There were no deaths or drug-related serious adverse events.
IgA nephropathy is a primary glomerulonephritis characterized by IgA deposition in the glomerular mesangium (also known as IgA nephritis or Berger's disease). Its pathogenesis is complex, with clinical manifestations including recurrent hematuria and proteinuria, potentially progressing to chronic renal failure. It is the most common primary glomerular disease globally, accounting for 30%-40% of cases in China and primarily affecting young adults aged 20-40. Prevalence and progression rates are higher in Asian populations compared to Western populations. It is estimated there are approximately 5 million patients in China, with over 100,000 new cases annually. The disease is often asymptomatic in early stages and progresses insidiously, with 20%-40% of patients progressing to end-stage renal disease within 20 years of diagnosis, requiring dialysis or kidney transplantation to sustain life.
Current treatment for IgA nephropathy primarily focuses on controlling blood pressure, reducing proteinuria, and delaying renal function decline. Commonly used drugs include angiotensin-converting enzyme inhibitors (ACEIs), angiotensin II receptor blockers (ARBs), corticosteroids, and immunosuppressants. However, these traditional therapies have limitations. For example, while corticosteroids can reduce inflammation to some extent, long-term use is associated with numerous side effects such as osteoporosis, elevated blood sugar, and increased infection risk. Furthermore, more effective treatment options are lacking for patients with severe disease or poor response to conventional therapies. Traditional treatments often only alleviate symptoms and are difficult to fundamentally halt disease progression.
Atacicept represents a new beacon of hope for patients with IgA nephropathy. It is a recombinant fusion protein containing the soluble transmembrane activator and CAML interactor (TACI) receptor, which can bind to B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL). These cytokines are members of the tumor necrosis factor family and promote B-cell survival and autoantibody production associated with certain autoimmune diseases, including IgA nephropathy and lupus nephritis. Vera Therapeutics believes atacicept, by targeting B cells and plasma cells to reduce autoantibodies, has the potential to be a best-in-class drug.
In January this year, Vera Therapeutics announced that the U.S. FDA had accepted its Biologics License Application (BLA) for atacicept for the treatment of adults with IgA nephropathy and granted it Priority Review. This application is primarily based on prespecified interim analysis data from the ORIGIN 3 study. Results showed that atacicept significantly reduced patients' proteinuria levels at week 36. The 24-hour urine protein-to-creatinine ratio (UPCR) decreased by 46% from baseline in the treatment group and showed a statistically significant and clinically meaningful 42% reduction compared to placebo (p<0.0001). Meanwhile, atacicept demonstrated a favorable safety profile across the ORIGIN program, generally comparable to placebo.
Baxdrostat, ulixacaltamide, and atacicept, targeting hypertension, essential tremor, and IgA nephropathy respectively—conditions with high unmet needs—have demonstrated promising clinical efficacy and safety. The successful development of these drugs not only provides patients with new treatment options and improves their quality of life but also reflects the pharmaceutical industry's attention and investment in niche disease areas. From a societal perspective, the emergence of these innovative therapies helps alleviate the medical burden on patients' families and society, enhancing patients' ability for self-care and social participation. Simultaneously, they inject new vitality into the biopharmaceutical field, encouraging more companies to conduct innovative research in the treatment of niche diseases and driving continuous progress across the industry.
06
Ending
A comprehensive review of the ten therapies discussed above reveals their common characteristics: deeper targeting of disease mechanisms, optimized administration strategies, and improved clinical response rates. These features signify that biopharmaceutical R&D is transitioning from a phase of "broad-spectrum inhibition" to a new era of "precision modulation." These potentially approvable therapies not only expand the treatment boundaries for key indications but also reflect a paradigm shift in innovative drug development toward a patient-centric, data-driven decision-making model. In the future, the verification of their long-term safety, accessibility, and real-world effectiveness will become critical dimensions for assessing their clinical value and industry impact. The year 2026 may serve as an important observation window for evaluating the outcomes of this transformation.
Source: Evaluate; Collation of Public Information